Developmental Disturbances of Oral and Paraoral Structures
 . Craniofacial Anomalies
. Craniofacial Anomalies . Developmental Disturbances of Jaws
. Developmental Disturbances of Jaws
 . Abnormalities of Dental Arch Relations
. Abnormalities of Dental Arch Relations
 . Developmental Disturbances of Lips and Palate
. Developmental Disturbances of Lips and Palate
 . Hereditary Intestinal Polyposis Syndrome
. Hereditary Intestinal Polyposis Syndrome
 . Developmental Disturbances of Oral Mucosa
. Developmental Disturbances of Oral Mucosa
 . Developmental Disturbances of Gingiva
. Developmental Disturbances of Gingiva
 . Developmental Disturbances of Tongue
. Developmental Disturbances of Tongue
 . Developmental Disturbances of Oral Lymphoid Tissue
. Developmental Disturbances of Oral Lymphoid Tissue
 . Developmental Disturbances of Salivary Glands
. Developmental Disturbances of Salivary Glands
 . Developmental Disturbances in Size of Teeth
. Developmental Disturbances in Size of Teeth
 . Developmental Disturbances in Shape of Teeth
. Developmental Disturbances in Shape of Teeth
 . Developmental Disturbances in Number of Teeth
. Developmental Disturbances in Number of Teeth
 . Developmental Disturbances in Structure of Teeth
. Developmental Disturbances in Structure of Teeth
 . Disturbances of Growth (Eruption) of Teeth
. Disturbances of Growth (Eruption) of Teeth . Fissural (Inclusion, Developmental) Cysts of Oral Region
. Fissural (Inclusion, Developmental) Cysts of Oral RegionCraniofacial Anomalies
• Combination of genes. A child may receive a particular combination of gene(s) from one or both parents, or there may be a change in the genes at the time of conception, which results in a craniofacial anomaly.
• Environmental. There is no data that shows a direct correlation between any specific drug or chemical exposure causing a craniofacial anomaly. However, any prenatal exposure should be evaluated.
• Folic acid deficiency. Folic acid is a B vitamin found in orange juice, fortified breakfast cereals, enriched grain products, and green, leafy vegetables. Studies have shown that women who do not take sufficient folic acid during pregnancy, or have a diet lacking in folic acid, may have a higher risk of having a baby with certain congenital anomalies, including cleft lip and/or cleft palate.
Some of the most common types of craniofacial anomalies (Table 1-1) include:
Table 1-1
Examples of most common craniofacial anomalies
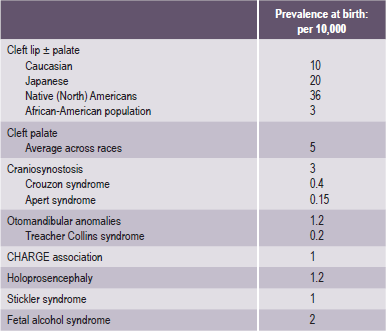
Source: Rovin et al, 1964; Temple, 1989; Cohen et al, 1992; Lewanda et al, 1992; Croen et al, 1996; Derijcle et al, 1996; Sampson et al, 1997; Blate et al, 1998.
• Cleft lip and/or cleft palate. A separation that occurs in the lip or the palate or both. Cleft lip and cleft palate are the most common congenital craniofacial anomalies seen at birth.
• Craniosynostosis. A condition in which the sutures in the skull of an infant close too early, causing problems with normal brain and skull growth. Premature closure of the sutures may also cause the pressure inside the head to increase and the skull or facial bones to change from a normal, symmetrical appearance.
• Hemifacial microsomia. A condition in which the tissues on one side of the face are underdeveloped, affecting primarily the ear (aural), mouth (oral), and jaw (mandibular) areas. Sometimes, both sides of the face can be affected and may involve the skull as well as the face. Hemifacial microsomia is also known as Goldenhar syndrome, brachial arch syndrome, facio-auriculovertebral syndrome (FAV), oculo-auriculovertebral spectrum (OAV), or lateral facial dysplasia.
• Vascular malformation. A birthmark or a growth, present at birth, which is composed of blood vessels that can cause functional or esthetic problems. Vascular malformations may involve multiple body systems. There are several different types of malformations, named after the type of blood vessel that is predominantly affected. Vascular malformations are also known as lymphangiomas, arteriovenous malformations, and vascular gigantism.
• Hemangioma. A type of birthmark; the most common benign (noncancerous) tumor of the skin. Hemangiomas may be present at birth (faint red mark) or appear in the first month after birth. A hemangioma is also known as a port wine stain, strawberry hemangioma, and salmon patch.
• Deformational (or positional) plagiocephaly. A misshapen (asymmetrical) shape of the head (cranium) from repeated pressure to the same area of the head. Plagiocephaly literally means ‘oblique head’ (from the Greek ‘plagio’ for oblique and ‘cephale’ for head).
Collectively they affect a significant proportion of the global society (Table 1-1).
Global Epidemiology
The frequency of occurrence of cleft lip, with or without cleft palate, has been computed on a global scale and is estimated to be 1 in every 800 newborn babies (Tables 1-2 and 1-3). A child is therefore born with a cleft somewhere in the world approximately every two-and-half minutes. Accurate data on the frequency of occurrence of these disorders is relevant for implementing strategies aimed at primary prevention and effective management of these disabled children (Table 1-2). Like anywhere else, the epidemiological data in this situation is also inherently handicapped by:
Table 1-2
Cleft lip with or without cleft palate
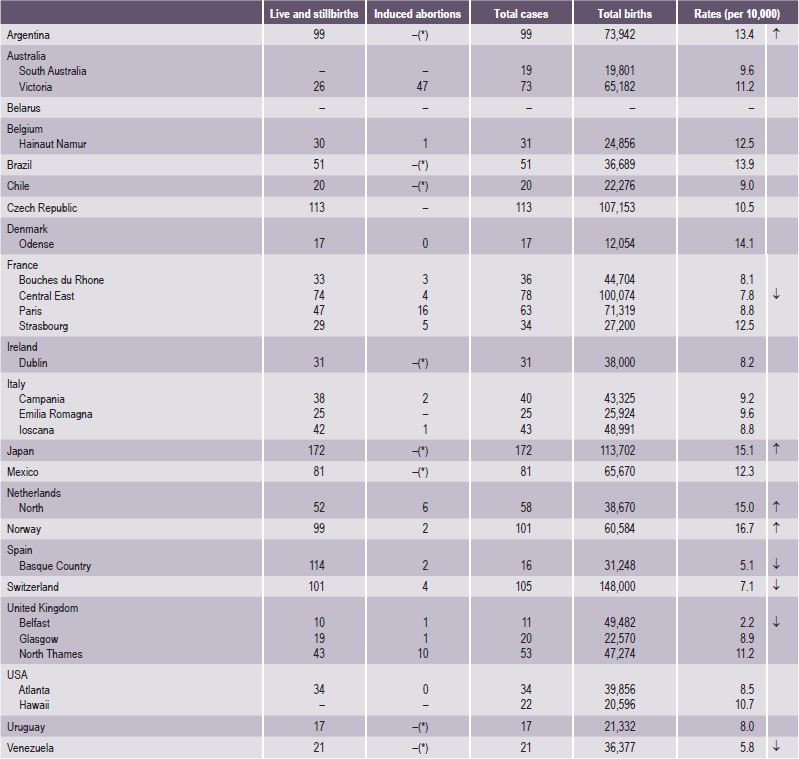
Source: WHO (1998), World Atlas of Birth Defects (1st Edition).
*Abortion for birth defect not permitted.
Table 1-3
Cleft palate without cleft lip
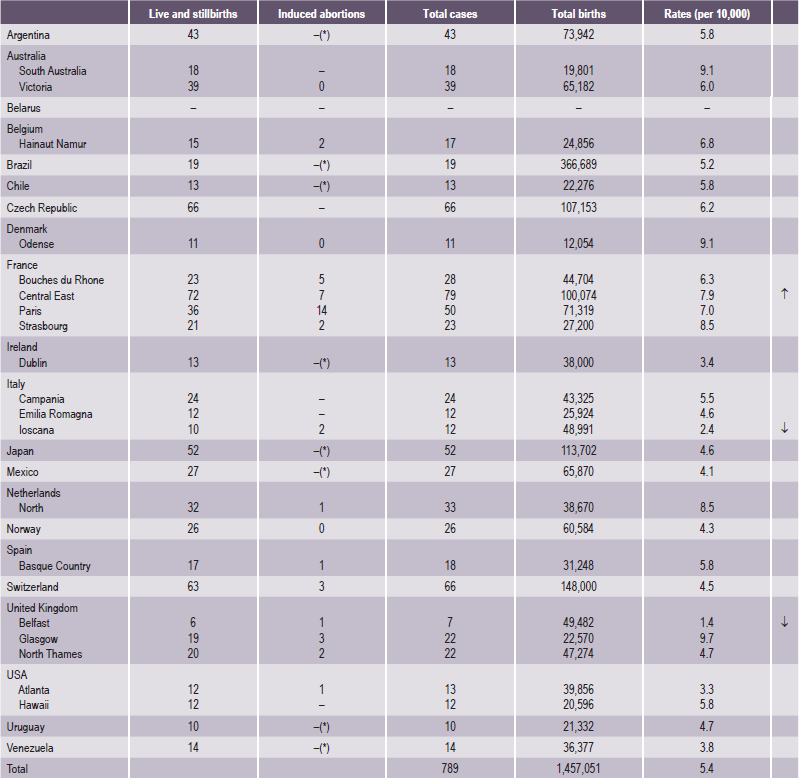
Source: WHO (1998), World Atlas of Birth Defects (1st Edition).
*Abortion for birth defect not permitted.
Registration of Targeted Craniofacial Anomalies in India
1. Three multicenter studies in India have provided almost similar frequency of CFA: meta-analysis of 25 early studies from 1960–1979, involving 407,025 births, showed:
CL/P = 440 cases, 1.08 per 1,000 births,
CP = 95 cases, 0.23 per 1,000 births.
2. A prospective national study of malformations in 17 centers from all over India from September 1989 to September 1990 involving 47,787 births showed:
CL/P = 64 cases, 1.3 per 1,000 births,
CP = 6 cases, 0.12 per 1,000 births.
3. The latest three-center study, conducted in 1994–1996, involved 94,610 births in Baroda, Delhi and Mumbai, and showed a frequency of:
This was the most rigorously conducted study and it found the number of infants born every year with CLP to be 28,600; this means 78 affected infants are born every day, or three infants with clefts are born every hour (Table 1-4)!
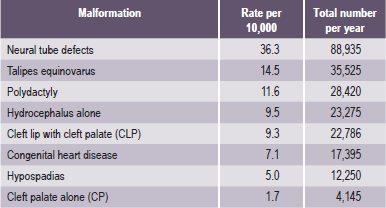
Another reason why a registry would be desirable is the changing pattern of morbidity and mortality in India emerging as a result of the achievements in immunization, the success in providing primary health care and the existence of a well-developed health infrastructure. In many university and city hospitals congenital malformations and genetic disorders have become important causes of illness. All these reasons show that starting a registry of these disorders deserves high priority in India.
Existing epidemiological data on CFA
• Higher frequency of CL+CP among Indian males is similar to that observed among Caucasians. The ratio is more than that observed in Africans and Japanese.
• The higher prevalence of CL+CP as compared with CL among Indians is like that observed in Africans, and is more than that observed in Caucasians.
• Children born prematurely are more frequently affected in India, as elsewhere.
• About 10.9% of 459 cases of all clefts are syndromic in Chennai. Of these, about 50 % are due to single-gene disorders, about 18% due to chromosomal disorders, and the rest due to undetermined causes.
• Chromosomal studies would be desirable in cases with associated abnormalities.
• Syndromes are more commonly associated with CP than with CL, as elsewhere.
• Lateralization (more clefts on the left side) in India is similar to that observed in other races.
• In one study in India, the intake of drugs was observed in 18% of parents — mostly steroidal compounds (progestogens as tests for pregnancy).
• A greater history of terminated pregnancies has been observed among cases, as compared with controls.
• History of severe vomiting has been observed to be about six times more common among case mothers than among controls.
• There is some difference in the frequency of orofacial clefts in different states in India; however, this needs verification. The state of origin (or mother tongue) of the parents should be recorded.
• Clefts are more commonly found in certain caste groups among Hindus.
• In India CP has less frequency in those with blood group A.
• CL occurs more in those with group O and AB.
• Association of clefts with certain HLA types has been documented in India.
• In a study in Chennai, significantly more consanguinity was observed among couples having children with clefts as compared with controls.
Genetics: Principles And Terminology
Genotype and Phenotype
The science of genetics is concerned with the inheritance of traits, whether normal or abnormal, and with the interaction of genes and the environment. This latter concept is of particular relevance to medical genetics, since the effects of genes can be modified by the environment.
Table 1-5
Genes involved in craniofacial and dental disorders, sorted by acronym gene name
| Acronym | Full name |
| ACTC | Actin, Alpha, Cardiac Muscle |
| APP | Amyloid Beta A4 Precursor Protein (APP) |
| ARVCF | Armadillo Repeat Gene Deleted in VCFS |
| ATP&E | ATPhase, H+ Transporting, Lysosomal, Subunit E |
| CA1 | Carbonic Anhydrase 1 |
| CLTCL1 | Clathrin, Heavy Polypeptide-Like 1 |
| COL01A1 | Collagen, Type I, Alpha-1 |
| COL01A2 | Collagen, Type I, Alpha-2 |
| COL02A1 | Collagen, Type II, Alpha-1 |
| COL04A4 | Collagen, Type IV, Alpha-4 |
| COL04A5 | Collagen, Type IV, Alpha-5 |
| COL05A1 | Collagen, Type V, Alpha-1 |
| COL06A1 | Collagen, Type VI, Alpha-1 |
| COL10A1 | Collagen, Type X, Alpha-1 |
| COMT | Catechol-O-Methyltransferase |
| CYLN2 | Cytoplasmic Linker 2 |
| DCN | Decorin |
| DGSI | DiGeorge Syndrome Critical Region Gene |
| ELN | Elastin |
| FBLN2 | Fibulin 2 |
| FBN1 | Fibrillin 1 |
| FGF8 | Fibroblast Growth Factor 8 |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FGFR2 | Fibroblast Growth Factor Receptor 2 |
| FGFR3 | Fibroblast Growth Factor Receptor 3 |
| GNAS1 | Guanine Nucleotide-Binding Protein, Alpha-Stimulating Activity Polypeptide |
| GOLGA1 | Golgi Autoantigen, Golgin Subfamily A, 1 |
| GP1BB | Glycoprotein Ib, Platelet, Beta Polypeptide |
| GPC3 | Glypican 3 |
| GPC4 | Glypican 4 |
| GPR1 | G Protein-Coupled Receptor 1 |
| GTF2I | General Transcription Factor II-I |
| HIP1 | Huntingtin-interacting Protein 1 |
| HIRA | Histone Cell Cycle Regulation Defective, S. Cerevisiae, Homolog of, A |
| HOXD10 | Homeo Box D10 |
| ITGB2 | Integrin, Beta-2 |
| KRT04 | Keratin 4 |
| KRT06A | Keratin 6A |
| KRT06B | Keratin 6B |
| KRT13 | Keratin 13 |
| KRT16 | Keratin 16 |
| KRT17 | Keratin 17 |
| LAMB1 | Laminin, Beta-1 |
| MEOX2 | Mesenchyme Homeo Box 2 |
| MSX1 | MSH, Drosophila, Homeo Box, Homolog of, 1 |
| MSX2 | MSH (Drosophila) Homeo Box Homolog 2 |
| PNUTL1 | Peanut-Like 1 |
| PTHR1 | Parathyroid Hormone Receptor 1 |
| RO60 | Autoantigen Ro/SSA, 60–KD |
| SCZD | Schizophrenia |
| SCZD4 | Schizophrenia 4 |
| SCZD8 | Schizophrenia 8 |
| SRC | V-SRC Avian Sarcoma (Schmidt-Ruppin A-2) Viral Oncogene |
| SRY | Sex-Determining Region Y |
| SSA1 | Sjögren Syndrome Antigen A1 |
| SSB | Sjögren Syndrome Antigen B |
| TNFRSF11B | Tumor Necrosis Factor Receptor Superfamily, Member 11B |
| TTPA | Tocopherol Transfer Protein, Alpha |
| TWIST | Twist, Drosophila, Homolog of |
| WBSCR1 | Williams-Beuren Syndrome Chromosome Region 1 |
Consideration of the heritability of a particular feature or trait requires a consideration of the relationship between genotype and phenotype. Genotype is defined as the genetic constitution of an individual, and may refer to specified gene loci or to all loci in general. An individual’s phenotype is the final product of a combination of genetic and environmental influences. Phenotype may refer to a specified character or to all the observable characteristics of the individual. The proportion of the phenotypic variance attributable to the genotype is referred to as heritability.
Genetic variation in man may be observed at two levels:
Morphological characters, on the other hand, such as the numerous dimensions used to describe the shape of the face and jaws, are furthest removed from the fundamental genetic level and are the end results of a vast complexity of interacting, hierarchical, biochemical, and developmental processes. Each gene is therefore likely to influence many morphological characters so that a deleterious mutation, although producing a unitary effect at the molecular level, almost always results in a syndrome of morphological abnormalities. When a gene is known to affect a number of different characters in this way its action is said to be pleiotropic. A reverse hierarchy also exists, making each morphological character dependent on many different genes (Mossey, 1999).
Modes of Inheritance
The exception to the rule that cells contain pairs of chromosomes applies to the gametes, sperm and ovum, which contain only single representatives of each pair of chromosomes, and therefore, of each pair of genes. When the two gametes join at fertilization, the new individual produced again has paired genes, one from the father and one from the mother. If a trait or disease manifests itself when the affected person carries only one copy of the gene responsible, along with one normal allele, the mode of inheritance of the trait is called dominant (Fig. 1-1A). If two copies of the defective gene are required for expression of the trait, the mode of inheritance is called recessive (Fig. 1-1B).
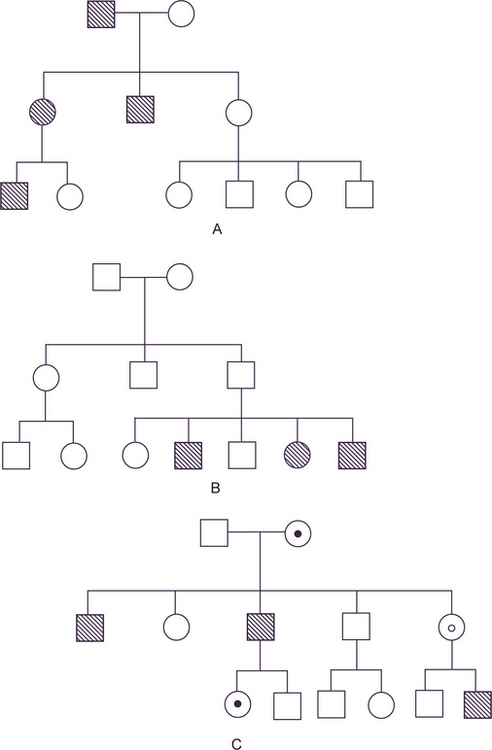
Figure 1-1 (A) Autosomal dominant pedigree. (B) Autosomal recessive pedigree. (C) X-linked recessive pedigree. Courtesy of PA Mossey, 1999.
The special case of genes carried on the X chromosome produces yet different pedigrees. Since male-to-male transmission is impossible and since females do not express the disease when they carry only one copy of the diseased gene (since it is modified by the homologous X chromosome), the usual pedigree consists of an affected male with clinically normal parents and children, but with affected brothers, maternal uncles, and other maternal male relatives (Fig. 1-1C). This mode of inheritance is described as X-linked recessive.
The determination of heritability for polygenic or multifactorial characters is difficult, as a feature of continuous variation is that different individuals may occupy the same position on the continuous scale for different reasons. Using mandibular length as an example, micrognathia can occur in chromosomal disorders, such as Turner’s syndrome, in monogenic disorders such as Treacher Collins syndrome or Stickler syndrome, or due to an intrauterine environmental problem, such as fetal alcohol syndrome. Combined with this the concept of etiological heterogeneity encompasses the principle of the same gene defect producing different phenotypic anomalies, and syndromes can be due to defective gene activity in different cells. Conversely, different gene defects or combinations of defective genes can produce a similar phenotypic abnormality. Genetic lethality or reduced reproductive fitness can also complicate the diagnostic picture and genomic imprinting can result in a gene defect ‘skipping’ a generation. These complexities serve to hamper progress in the understanding of polygenic or multifactorial disorders such as orofacial clefting (Mossey, 1999).
Multifactorial Inheritance
In contrast to single-gene inheritance, either autosomal or sex-linked, the pedigree pattern does not afford a diagnosis of multifactorial inheritance. In multifactorial traits, the trait is determined by the interaction of a number of genes at different loci, each with a small, but additive effect, together with environmental factors (i.e., the genes are rendering the individual unduly susceptible to the environmental agents). Many congenital malformations (Table 1-6) and common diseases of adult life are inherited as multifactorial traits and these are categorized as either continuous or discontinuous.
Table 1-6
Extrinsic
Mechanical
Unstretched uterine and abdominal muscles
Small maternal size
Amniotic tear
Unusual implantation site
Uterine leiomyomas
Unicornuate uterus
Bicornuate uterus
Twin fetuses
Intrinsic
Malformational
Spina bifida
Other central nervous system malformations
Bilateral renal agenesis
Severe hypoplastic kidneys
Severe polycystic kidneys
Urethral atresia
Functional
Neurologic disturbances
Muscular disturbances
Connective tissue defects
Molecular Genetics in Dental Development
The first sign of tooth development is a local thickening of oral epithelium, which subsequently invaginates into neural crest derived mesenchyme and forms a tooth bud. Subsequent epithelial folding and rapid cell proliferation result in first the cap, and then the bell stage of tooth morphogenesis. During the bell stage, the dentine producing odontoblasts and enamel secreting ameloblasts differentiate. Tooth development, like the development of all epithelial appendages, is regulated by inductive tissue interactions between the epithelium and mesenchyme (Thesleff, 1995).
There is now increasing evidence that a number of different mesenchymal molecules and their receptors act as mediators of the epithelial-mesenchymal interactions during tooth development. Of the bone morphogenetic proteins (BMPs) 2, 4, and 7 mRNAs shift between the epithelium and mesenchyme in the regulation of tooth morphogenesis (Aberg et al, 1997). The fibroblast growth factor (FGF) family have also been localized in epithelial and mesenchymal components of the tooth by immunohistochemistry (Cam et al, 1992); and in dental mesenchyme tooth development and shape is regulated by FGF8 and FGF9 via downstream factors MSX1 and PAX9 (Kettunen and Thesleff, 1998).
Control of Tooth Development
Homeobox genes have particular implications in tooth development. Muscle specific homeobox genes Msx-1 and Msx-2 appear to be involved in epithelial mesenchymal interactions, and are implicated in craniofacial development, and in particular, in the initiation developmental position (Msx-1) and further development (Msx-2) of the tooth buds (MacKenzie et al, 1991; Jowett et al, 1993). Further evidence of the role of Msx-1 comes from gene knock-out experiments which results in disruption of tooth morphogenesis among other defects (Satokata and Maas, 1994). Pax-9 is also transcription factor necessary for tooth morphogenesis (Neubuser et al, 1997). BMPs are members of the growth factor family (TGF) and they function in many aspects of craniofacial development with tissue specific functions. BMPs have been found to have multiple roles not only in bone morphogenesis, (BMP 5, for example, induces endochondral osteogenes is in vivo), but BMP 7 appears to induce dentinogenesis (Thesleff, 1995).
Congenital Deformations Of Head And Neck
Teratogenic Agents
Teratogens are agents that may cause birth defects when present in the fetal environment. Included under such a definition are a wide array of drugs, chemicals, and infectious, physical, and metabolic agents that may adversely affect the intrauterine environment of the developing fetus. Such factors may operate by exceedingly heterogeneous pathogenetic mechanisms to produce alterations of form and function as well as embryonic and/or fetal death (Gorlin et al, 1990).
It is of considerable interest to the dental profession that experimental studies of teratogenic agents have almost invariably revealed a variety of head, neck and oral malformations. Conway and Wagner have compiled a list of the most common malformations of the head and neck as shown in Table 1-7.
Table 1-7
| Type | Occurrence (%) |
| Unilateral incomplete | 33 |
| Unilateral complete | 48 |
| Bilateral incomplete | 7 |
| Bilateral complete | 12 |
Source: V Veau: Division palatine; anatomie, chirurgie, phonetique. Paris, Masson et Cie, 1931.
Stem cells are clonogenic cells that have the capacity for self-renewal and multilineage differentiation. The microenvironment in which stem cells reside is called a stem cell niche and is composed of heterologous cell types, extracellular matrix and soluble factors to support the maintenance and self-renewal of the stem cells. Stem cells have been isolated and characterized from a wide variety of sources. Adult progenitor cellular populations have been isolated from distinctive tissues and fluids, such as bone marrow, peripheral blood, umbilical cord blood, Wharton’s jelly, placenta, amniotic fluid and membrane, adipose tissue, dermis, hair follicles, synovial membrane, skeletal muscle, central nervous system, olfactory bulb, retina, and liver among others. From these biological sources, progenitors of the mesenchymal, epithelial, hematopoetic (discussed elsewhere), neural, endothelial and trophoblastic lineages have been identified (Fig. 1-2).
Figure 1-2 Mandibular micrognathia. Courtesy: Prof Anil Sukumaran, Prof R Rajendran. King Saud University, Riyadh, KSA.
There are two major categories of stem cells, which are of relevance to dentistry:
Developmental Disturbances of Jaws
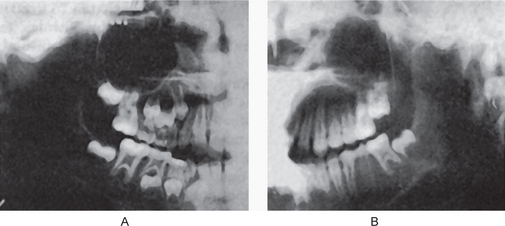
Micrognathia
The clinical appearance of mandibular micrognathia is characterized by severe retrusion of the chin, a steep mandibular angle, and a deficient chin button (Fig. 1-3).
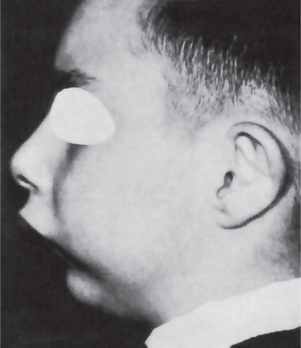
Micrognathia may be caused by or may be a feature of several conditions (Table 1-8).
Table 1-8
Congenital conditions
• Catel-Manzke syndrome
• Cerebrocostomandibular syndrome
• Cornelia de Lange syndrome
• Femoral hypoplasia—unusual facies syndrome
• Fetal aminopterin-like syndrome
• Miller-Dieker syndrome
• Nager acrofacial dysostosis
• Pierre Robin syndrome
• Schwartz-Jampel-Aberfeld syndrome
• van Bogaert-Hozay syndrome
Intrauterine acquired conditions
• Syphilis, congenital
Chromosomal abnormalities
• 49, XXXXX syndrome
• Chromosome 8 recombinant syndrome
• Cri du chat syndrome 5p
• Trisomy 18
• Turner’s syndrome
• Wolf-Hirschhorn syndrome
Mendelian inherited conditions
• CODAS (cerebral, ocular, dental, auricular, skeletal) syndrome
• Diamond-Blackfan anemia
• Noonan’s syndrome
• Opitz-Frias syndrome
Autosomal dominant conditions
• Camptomelic dysplasia
• Cardiofaciocutaneous syndrome
• CHARGE syndrome
• DiGeorge’s syndrome
• Micrognathia with peromelia
• Pallister-Hall syndrome
• Treacher Collins-Franceschetti syndrome
• Trichorhinophalangeal syndrome type 1
• Trichorhinophalangeal syndrome type 3
• Wagner vitreoretinal degeneration syndrome
Autosomal recessive conditions
• Bowen-Conradi syndrome
• Carey-Fineman-Ziter syndrome
• Cerebrohepatorenal syndrome
• Cohen syndrome
• Craniomandibular dermatodysostosis
• De la Chapelle dysplasia
• Dubowitz syndrome
• Fetal akinesia-hypokinesia sequence
• Hurst’s microtia-absent patellae-micrognathia syndrome
• Kyphomelic dysplasia
• Lathosterolosis
• Lethal congenital contracture syndrome
• Lethal restrictive dermopathy
• Marden-Walker syndrome
• Orofaciodigital syndrome type 4
• Postaxial acrofacial dysostosis syndrome
• Rothmund-Thomson syndrome
• Smith-Lemli-Opitz syndrome
• ter Haar syndrome
• Toriello-Carey syndrome
• Weissenbacher-Zweymuller syndrome
• Yunis-Varon syndrome
X-linked inherited conditions
• Atkin-Flaitz-Patil syndrome
• Coffin-Lowry syndrome
• Lujan-Fryns syndrome
• Otopalatodigital syndrome type 2
Autoimmune conditions
• Juvenile chronic arthritis
Source: www.diseasesdatabase.com, Health on the Net Foundation, 2005.
Macrognathia
• Paget’s disease of bone, in which overgrowth of the cranium and maxilla or occasionally the mandible occurs,
• Acromegaly, in which there is progressive enlargement of the mandible owing to hyperpituitarism in the adult, or
• Leontiasis ossea, a form of fibrous dysplasia in which there is enlargement of the maxilla.
Cases of mandibular protrusion or prognathism, uncomplicated by any systemic condition, are a rather common clinical occurrence (Fig. 1-4). The etiology of this protrusion is unknown, although some cases follow hereditary patterns. In many instances the prognathism is due to a disparity in the size of the maxilla in relation to the mandible. In other cases the mandible is measur-ably larger than normal. The angle between the ramus and the body also appears to influence the relation of the mandible to the maxilla, as does the actual height of the ramus. Thus prognathic patients tend to have long rami which form a less steep angle with the body of the mandible. The length of the ramus, in turn, may be associated with the growth of the condyle. It may be reasoned, therefore, that excessive condylar growth predisposes to mandibular prognathism.
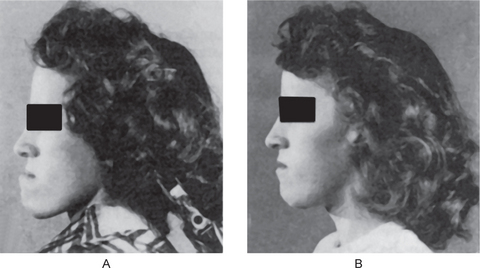
Figure 1-4 Macrognathia (prognathia) of the mandible. (A) The protrusion of the mandible is obvious. (B) The same patient after surgical correction (ostectomy) (Courtesy of Dr G Thaddeus Gregory and Dr J William Adams.
Facial Hemihypertrophy (Hyperplasia)
Hemihyperplasia is a rare developmental anomaly characterized by asymmetric overgrowth of one or more body parts. Although the condition is known more commonly as hemihypertrophy, it actually represents a hyperplasia of the tissues rather than a hypertrophy. Hemihyperplasia can be an isolated finding, but it also may be associated with a variety of malformation syndromes (Table 1-9). Almost all cases of isolated hemihyperplasia are sporadic.
Table 1-9
Malformation syndromes associated with hemihyperplasia
• Beckwith-Wiedemann syndrome
• Neurofi bromatosis
• Klippel-Trenaunay-Weber syndrome
• Proteus syndrome
• McCune-Albright syndrome
• Epidermal nevus syndrome
• Triploid/diploid mixoploidy
• Langer-Giedion syndrome
• Multiple exostoses syndrome
• Maffucci’s syndrome
• Ollier syndrome
• Segmental odontomaxillary dysplasia
Hoyme et al (1998) provided an anatomic classification of hemihyperplasia:
• Complex hemihyperplasia is the involvement of half of the body (at least one arm and one leg); affected parts may be contralateral or ipsilateral,
• Simple hemihyperplasia is the involvement of a single limb, and
• Hemifacial hyperplasia is the involvement of one side of the face.
Oral Manifestations
Characteristically, the permanent teeth on the affected side develop more rapidly and erupt before their counterparts on the uninvolved side (Fig. 1-5). Coincident to this phenomenon is premature shedding of the deciduous teeth. The bone of the maxilla and mandible is also enlarged, being wider and thicker, sometimes with an altered trabecular pattern.
Treatment and Prognosis
There is no specific treatment for this condition other than attempts at cosmetic repair. Cosmetic surgery is advised after cessation of growth. Effect on life expectancy is not certain, but in some cases patients have lived a normal life span. Periodic abdominal ultrasound/ MRI is recommended to rule out tumors.
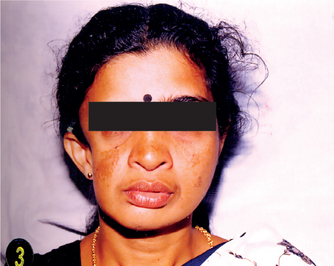
Facial Hemiatrophy (Parry-Romberg syndrome, Romberg-Parry syndrome, progressive facial hemiatrophy, progressive hemifacial atrophy)
Clinical Features
The disease occurs more frequently in women; female to male ratio is 3 : 2. It has a slight predilection for the left side and appears in the first or second decades of life. It progresses over a period of two and 10 years, and atrophy appears to follow the distribution of one or more divisions of the trigeminal nerve. The resulting facial flattening may be mistaken for Bell’s palsy (Fig. 1-6).
Abnormalities of Dental Arch Relations
Class I. Arches in normal mesiodistal relations 69.0%.
Class II. Mandibular arch distal to normal in its relation to the maxillary arch.
Division 1. Bilaterally distal, protruding maxillary incisors 9.0.
Subdivision. Unilaterally distal, protruding maxillary incisors 3.5.
Division 2. Bilaterally distal, retruding maxillary incisors 4.0.
Subdivision. Unilaterally distal, retruding maxillary incisors 10.0.
Class III. Mandibular arch mesial to normal in its relation to the maxillary arch.
Developmental Disturbances of Lips And Palate
Congenital Lip and Commissural Pits and Fistulas
Clinical Features
The lip pit or fistula is a unilateral or bilateral depression or pit that occurs on the vermilion surface of either lip but far more commonly on the lower lip (Fig. 1-7A). In some cases a sparse mucous secretion may exude from the base of this pit. The lip sometimes appears swollen, accentuating the appearance of the pits.
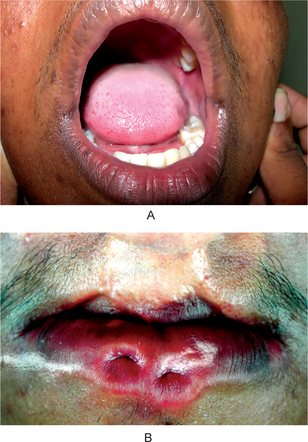
Figure 1-7 (A) Congenital lip pits. (B) Congenital commissural pits. Courtesy of Dr Spencer Lilly, Meenakshi Ammal Dental College, Chennai.
Commissural pits appear as unilateral or bilateral pits at the corners of the mouth on the vermilion surface (Fig. 1-7B). An actual fistula may be present from which fluid may be expressed. Whether this tract, either in lip or commissural fistulas, represents a true duct is not clear. Interestingly, in several cases preauricular pits have been reported in association with commissural pits.
van der Woude Syndrome (Cleft lip syndrome, lip pit syndrome, dimpled papillae of the lip)
Clinical Features
In general, van der Woude syndrome affects about 1 in 100,000–200,000 people. About 1–2% of patients with cleft lip or palate have van der Woude syndrome. The van der Woude syndrome affects both genders equally and no difference among them have been reported. The severity of the van der Woude syndrome varies widely, even within families. About 25% of individuals with the van der Woude syndrome have no findings or minimal ones, such as missing teeth or trivial indentations in the lower lips. Others have severe clefting of the lip or palate. The hallmark of the van der Woude syndrome is the association of cleft lip and/or palate with distinctive lower lip pits. This combination is seen in about 70% of those who are overtly affected but in less than half of those who carry the gene. The cleft lip and palate may be isolated. They may take any degree of severity and may be unilateral or bilateral. Submucous cleft palate is common and may be easily missed on physical examination. Hypernasal voice and cleft or bifid uvula are clues to this diagnosis. It is possible as well that a bifid uvula is an isolated finding in certain individuals with the van der Woude syndrome. The lower lip pits seen in this syndrome are fairly distinctive (Fig. 1-8). The pits are usually medial, on the vermilion portion of the lower lip. They tend to be centered on small elevations in infancy, but are simple depressions in adults. These pits are often associated with accessory salivary glands th/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


