Diseases of the Nerves and Muscles
 . Diseases of the Nerves
. Diseases of the Nerves . Disturbances of Fifth Cranial Nerve
. Disturbances of Fifth Cranial Nerve . Disturbances of Seventh Cranial Nerve
. Disturbances of Seventh Cranial Nerve . Disturbances of Ninth Cranial Nerve
. Disturbances of Ninth Cranial Nerve . Miscellaneous Disturbances of Nerves
. Miscellaneous Disturbances of Nerves . Diseases of the Muscles
. Diseases of the Muscles . Dystrophies
. Dystrophies . Myotonias
. Myotonias . Myasthenias
. Myasthenias . Myositis
. Myositis . Heterotopic Ossification
. Heterotopic Ossification . Proliferative Myositis
. Proliferative Myositis . Miscellaneous Myopathies
. Miscellaneous MyopathiesDiseases of the Nerves
Disturbances of Fifth Cranial Nerve
Trigeminal Neuralgia (Tic douloureux, trifacial neuralgia, Fothergill’s disease)
Clinical Features
Older adults are more commonly affected by trigeminal neuralgia than young persons, the disease seldom occurring before the age of 35 years. Females are more commonly affected (3:2). It is a well-established fact, but a completely unexplained one, that the right side of the face is affected in more patients than the left by a ratio of about 1.7:1.
Differential Diagnosis
Treatment
One of the earliest forms of treatment was peripheral neurectomy—sectioning of the nerve at the mental foramen, or at the supraorbital or infraorbital foramen. Since any relief afforded is temporary, this form of treatment has not been extensively used in recent years. The injection of alcohol either into a peripheral nerve area or centrally into the gasserian ganglion has had many proponents throughout the years, despite its temporary benefit and attendant dangers. The patient may experience respite from all symptoms for a period of six months to several years after alcohol injection. The injection of boiling water into the gasserian ganglion has also been reported to be beneficial in causing respite from pain. Surgical sectioning of the trigeminal sensory root by any of a number of techniques has come to be recognized by many surgeons as the treatment of choice when attempting to obtain a permanent cure.
Sphenopalatine Neuralgia (Sphenopalatine ganglion neuralgia, lower-half headache, Sluder’s headache, vidian nerve neuralgia, atypical facial neuralgia, histamine cephalgia, Horton’s syndrome, cluster headache, periodic migrainous neuralgia)
Treatment
Numerous methods of treatment of the sphenopalatine ganglion syndrome have been proposed, none of which is successful in every instances. One of the most widely used of these has been cocainization of the sphenopalatine ganglion or alcohol injection of this structure. Resection of the ganglion has been carried out in some instances, as well as surgical correction of septal defects. It has been found that ergotamine will often produce immediate and complete relief of symptoms. In those cases where it is not totally effective, combining it with methysergide, an antiserotonin agent, appears to produce a synergistic action usually providing total relief. However, both drugs carry some risk of serious side effects if given in large doses or over a prolonged period. Invasive nerve blocks and ablative neurosurgical procedures all have been implemented successfully in refractory cases.
Orolingual Paresthesia (Glossodynia or painful tongue, glossopyrosis or ‘trigger zone’ tongue)
Etiology
• Deficiency states such as pernicious anemia and pellagra
• Gastric disturbances such as hyperacidity or hypoacidity
• Referred pain from abscessed teeth or tonsils
• Oral habits such as excessive use of tobacco, spices, and the like
• Local dental causes such as dentures, irritating clasps or new fixed bridges.
In addition, Shafer pointed out two other possible etiologic factors: an electrogalvanic discharge occurring between dissimilar metallic dental restorations, and temporomandibular joint disturbances.
Disturbances of Seventh Cranial Nerve
Bell’s Palsy (Seventh nerve paralysis, facial paralysis)
Etiology
Bell’s palsy is considered an idiopathic facial paralysis; however, an infectious cause has been reported. Herpes simplex virus (HSV) has been isolated in many patients with Bell’s palsy and is most likely the infectious agent. There is more than one etiologic agent with a shared common pathway leading to facial neuropathy. Actual pathophysiology is unknown. A popular theory proposes that the inflammation of the facial nerve with resultant edema causes nerve compression while it passes through the temporal bone. Various inflammatory, demyelinating, ischemic, or compressive processes may impair neural conduction at this unique anatomic site.
Clinical Features
The muscular paralysis manifests itself by the drooping of the corner of the mouth, from which saliva may run, the watering of the eye, and the inability to close or wink the eye, which may lead to infection. When the patient smiles, the paralysis becomes obvious, since the corner of the mouth does not rise nor does the skin of the forehead wrinkle or the eyebrow raise (Fig. 20-1). The patient has a typical mask like or expressionless appearance. Speech and eating usually become difficult, and occasionally the taste sensation on the anterior portion of the tongue is lost or altered.
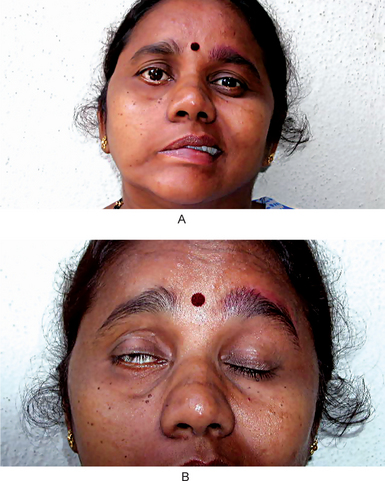
Figure 20-1(A, B) Bell’s palsy due to facial nerve paralysis.
The patient demonstrates the typical unilateral paralysis of the facial musculature with inability to smile or close the eye on the affected side Courtesy of Dr Ajayprakash, Department of Oral Pathology, Kamineni Institute of Dental Sciences, Andhra Pradesh.
Disturbances of Ninth Cranial Nerve
Glossopharyngeal Neuralgia
Treatment
The treatment of glossopharyngeal neuralgia has generally consisted in resection of the extracranial portion of the nerve or intracranial section. The injection of alcohol into the glossopharyngeal nerve has not been as widely accepted as has similar treatment in the case of trigeminal neuralgia. Periods of remission with subsequent recurrence are common in this disease.
Miscellaneous Disturbances of Nerves
Multiple Sclerosis (Disseminated sclerosis)
Clinical Features
• A variety of ocular disturbances, including visual impairment as a manifestation of retrobulbar neuritis, nystagmus and diplopia.
• Fatigability, weakness and stiffness of extremities with ataxia or gait difficulty involving one or both legs.
• Superficial or deep paresthesia.
• Personality and mood deviation toward friendliness and cheerfulness.
• Autonomic effector derangements, such as bladder and/or rectal retention or incontinence.
Charcot’s triad is a well-known diagnostic triad characteristic of multiple sclerosis but not invariably present. It consists of intention tremor, nystagmus and dysarthria or scanning speech, an imperfect speech articulation.
Ménière’s Disease (Ménière’s disease, Ménière’s syndrome, endolymphatic hydrops)
Migraine (Migraine syndrome)
Etiology
The mechanisms of migraine are not completely understood. However, the advent of new technologies has allowed formulation of current concepts that may explain parts of the migraine syndrome. For many years, headache during a migraine attack was thought to be a reactive hyperemia in response to vasoconstriction-induced ischemia during aura (Fig. 20-2). This explained the throbbing quality of the headache, its varied localization, and the relief obtained from ergots; however, it does not explain the prodrome and associated features.
Temporal/Giant Cell Arteritis
Treatment and Prognosis
The response of temporal arteritis to corticosteroid therapy is excellent, and clinical manifestations subside within a few days. In occasional cases in which there is widespread systemic vascular involvement, the course of the disease may be progressively downhill and may terminate fatally.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


