Abstract
Many advances in healthcare are built on advances in technology. In the case of fetal medicine, technology has availed an entirely new patient population. The authors report a case of severe micrognathia and Pierre Robin Sequence that was diagnosed prenatally. Antenatal planning and treatment were instituted via the Fetal Diagnosis/Treatment Team to avoid loss of the neonate’s airway. An EXIT procedure was utilized to ensure a secure airway. The benefits of team care for these types of deformities are highlighted including the importance of craniomaxillofacial specialists.
Encountering the fetus as a patient has become a reality. Advances in prenatal imaging allow us to see and diagnose abnormalities not previously appreciated. As these technological advances continue, surgeons are called upon to provide expertise in the diagnosis and treatment of anomalies in the prenatal, intra-partum and immediate post-partum periods . Clinicians can plan for the delivery of the neonate in such a way that anomalies are optimally managed, and the adverse impact on the neonate’s health minimized. Advances in imaging have provided an entirely new patient population.
Abnormalities of the craniomaxillofacial structures can have an immediate effect on the viability of a neonate because of their impact on the airway. Procedures such as ex-utero intra-partum treatment (EXIT) and extracorporeal mechanical oxygenation (ECMO) have been developed to address these issues . Other anomalies that are not an immediate threat to the airway at birth, may require an early management protocol during the first days or weeks of life. Others, such as clefting, may require a staged reconstruction over years, and planning is essential for the best outcome. The decision making process regarding treatment is best accomplished in an interdisciplinary forum.
The authors report a case of severe micrognathia and Pierre Robin Sequence (PRS) that was diagnosed prenatally. Antenatal planning and treatment were instituted via the Fetal Diagnosis and Treatment Team to avoid a potentially disastrous airway outcome. An EXIT procedure was utilized to ensure a secure airway. The benefits of inter-disciplinary team care for these types of deformities are highlighted including the importance of craniomaxillofacial specialists as an integral part of this team.
Case report
A 21-year-old Gravida 1 Para 0 with an unremarkable health history, with a fetus at the gestational age of 20.33 weeks, was referred to the Fetal Diagnosis Treatment Team due to an elevated maternal serum alpha-fetoprotein (AFP) of 2.67 multiples of the median. A genetics consultation was obtained as was an ultrasound (US) which showed evidence of micrognathia ( Fig. 1 ). An amniocentesis was completed demonstrating a normal karyotype and a normal amniotic fluid level of AFP. Further review revealed a significantly hypoplastic mandible compared with age-matched control data. The patient was asked to return for a follow-up US because of the fetal dysmorphology.
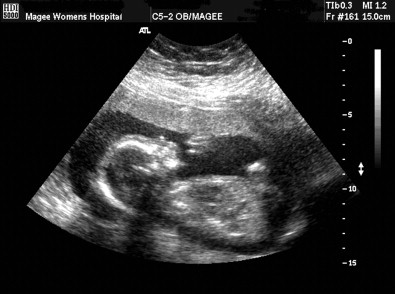
At 30.3 weeks’ gestation, an US ( Fig. 2 ) was repeated with more detail to evaluate the fetal anatomy and assess growth. It showed poor fetal weight gain (twenty-ninth percentile for gestational age), polyhydramnios and micrognathia. At a gestational age of 34.6 weeks, a follow-up US was carried out and showed declining growth (fourteenth percentile) and polyhydramnios. Twice weekly antenatal surveillance was begun.

Given the suspected fetal abnormalities and polyhydramnios, the decision was made to deliver after amniocentesis for fetal lung maturity at 37 weeks’ gestation. At 37.3 weeks’ gestation, the patient returned for ultrasound and amniocentesis, and due to extreme intrauterine growth restriction (less than the fifth percentile) the decision was made to deliver. Members of the team were assembled to prepare for the delivery and perform an EXIT procedure to secure an airway if necessary.
Owing to a variety of factors including maternal habitus, placental location and breech presentation of the fetus, the team felt that a classic EXIT procedure could not be performed. The team thought that the umbilical cord could be left attached after the c-section and would be able to provide oxygenation to the fetus for 1–2 min, after which the cord would have to be transected and a definitive airway established. The patient underwent a c-section and the baby was delivered and turned to lie over the mother’s right thigh with the umbilical cord intact utilizing a modified EXIT procedure. Direct laryngoscopy was attempted, but due to the severe micrognathia it was unsuccessful. The baby was demonstrating signs of upper airway obstruction and given the inability to maintain oxygenation via uteroplacental circulation, a tracheostomy was performed through a vertical midline incision. After dissection and identification of the trachea, another vertical midline incision was made through the second and third tracheal rings and a 3.0 Neonatal Shiley was inserted.
The diagnosis of Cornelia de Lange syndrome was made based on the phenotype ( Fig. 3 ). An evaluation directed at the expected deformities associated with Cornelia de Lange syndrome was begun. Long-term airway management was planned including distraction osteogenesis to attempt an improvement of airway dynamics.

Discussion
When a fetus presents with anomalies, delivery of the child with coordinated management is facilitated in a team setting. The antenatal diagnosis of a malformation, lesion or syndrome carries with it interlinked social, ethical and medical issues. These circumstances bring complex care issues, stress and anxiety, and tax the coping skills of any family. The Fetal Diagnosis/Treatment Team functions as a resource to aid them throughout the process.
Anomalies that impact the airway, such as micrognathia, congenital diaphragmatic hernias (CDH), congenital high airway obstruction syndrome (CHAOS), congenital cystic adenomatoid malformation (CCAM), and giant cervical masses (most commonly cervical teratoma and cystic hygroma) can be planned for prior to delivery to avoid emergency intubations or potential fatal outcomes. These anomalies are often amenable to the EXIT procedure. The EXIT procedure was originally designed for use in reversing tracheal occlusion in CDH where in-utero tracheal clips were placed to induce fetal lung development, also known as the PLUG phenomenon (plug the lung until grows) . Input from multiple disciplines is important for achieving a good outcome . It is in this realm that surgeons with experience of craniofacial anomalies are finding their niche.
Having characteristics of the anomaly imaged prior to intervention is essential. Two- and three-dimensional ultrasound (two-dimensional images converted to volumetric sequences) are valuable modalities for the identification of fetal structural abnormalities . Imaging studies are also important for guiding the maternal-fetal medicine specialist during amniocentesis or other fetal interventions. Fetal MRI (single shot fast spin echo T2 weighted images), have defined these anomalies in great detail .
The EXIT procedure was designed for use in reversing tracheal occlusion in CDH where in-utero tracheal clips were placed to induce fetal lung development . The EXIT procedure provides sufficient oxygenation to the fetus via ‘maternal-fetal bypass’ while the cervical mass or obstruction is resected allowing access to the airway, or an airway is established through oral endotracheal intubation, or tracheostomy. Resection of a lesion or other definitive management can then be performed in a controlled setting. Early reports have documented that this technique can be maintained for up to an hour as demonstrated through normal cord blood gas values. A resection of a giant cervical teratoma that lasted for two and a half hours has been reported, without adverse events to mother or child .
Fetal-medical clinicians and pediatric craniomaxillofacial surgeons have expanded the scope of these procedures to include EXIT to ECMO, EXIT to resection, and EXIT to separation strategy in conjoined twins. Hirose et al. in a 10 year retrospective study of all patients who underwent an EXIT procedure for neck masses (a total of 5) reported that tracheostomy was performed in three, oral intubation was successful in one and the last underwent resection while on placental support . Elective airway management was used, and all patients were treated successfully without adverse effects.
Micrognathia, whether as an isolated condition or as part of a defined syndrome, can present with significant airway and feeding difficulties. It is becoming more common for ultrasonographers to diagnose a significantly hypoplastic mandible or other craniofacial anomalies during routine screening examinations. Visualizing the mandible is technique sensitive and requires a skilled ultrasonographer with adequate experience to visualize the mandible in the true sagittal plane. Incomplete and para-sagittal views may result in a false-positive for hypoplasia of the mandible even when using three-dimensional sweeping techniques. The diagnosis of a hypoplastic mandible should alert clinicians to consider other anomalies that may be associated with a syndromic diagnosis (i.e. Cornelia DeLange Syndrome, Sticker Syndrome).
Cornelia de Lange syndrome occurs in several forms and is an autosomal dominant disorder with most cases being sporadic. There are three variants based on three gene loci. The present patient was diagnosed with Type I (the more common type), which is caused by a mutation in the 5p13.1 Nipped B-like gene (NIPBL). It occurs in approximately 1 in 100,000 births. Type II is X-linked and occurs due to a defect at the Xp11.22–p11.21 gene loci, and type III occurs due to a mutation at the SMC3 gene (10q25). It is one of the many syndromes that can have Pierre Robin Sequence associated with its other features. Major features include prenatal growth retardation, short stature, microcephaly, a long philtrum, low-set ears, long and curly eyelashes, short vertical nasal height, cleft lip and palate, congenital heart defects, duplication of bowel, renal anomalies, hirsutism with a mono-brow, mental retardation, hypertonicity, and self-injurious behavior. The expression in each of the types is variable. Prenatal testing for each of the types is possible, but expensive. It rarely makes an outcome difference for surgical planning if the disorder is recognized prenatally as a specific syndrome, except to note any associated defects such as cardiac anomalies. The key aspect of management of patients with Cornelia de Lange syndrome is appreciating the degree of dysmorphology and severity of the mandibular hypoplasia and to anticipate airway compromise.
A hypoplastic mandible can result in retro-placement of the tongue and floor of mouth resulting in airway obstruction that may require immediate intervention. Management of airway obstruction associated with micrognathia is generally tiered based on severity. The severity of micrognathia can be assessed prenatally during ultrasound examination utilizing measurements from age-matched data. When hypoplasia of the mandible is noted prenatally and the micrognathia is severe, the team may consider options including an EXIT procedure ( Fig. 4 ). Maternal circulation can be sustained in the early moments after birth to assess whether intubation is possible, or if tracheostomy is required. The long-term airway management can then be considered. This may include tracheostomy, early distraction osteogenesis, or other techniques based on a thorough airway evaluation. If survivable micrognathia is discovered after birth, mild cases may be managed with positional therapy and prone feedings. More significant cases may benefit from temporary nasal stenting, tongue–lip adhesion, distraction osteogenesis, or tracheostomy.



