These are exciting times in the field of pain research. Every day brings advances in our understanding of pain mechanisms, and with each new advancement there is hope that these findings will lead to the development of novel and more effective analgesics not only for acute pain, but also for the more difficult and challenging to manage chronic pain conditions. The field of pain research represents an evolving field, where early studies identified basic pain pathways and the characterization of different fiber types and receptors that were activated by noxious stimuli. With this basic knowledge, the receptors and transmitters involved in the activation and inhibition of these different pathways were identified, and significant changes in their expressions were seen after inflammatory and nerve lesions. These changes in receptors and transmitters were also correlated with the increased activity of pain pathways in pathologic conditions. Advances in brain imaging techniques have led to the concept of pain as a widely distributed system involving many different nervous system structures that represent the affective and sensory aspects of the pain experience. Molecular approaches are being used to map the intricacies of the intracellular signaling pathways that are activated when molecules bind to a receptor or channels open in response to specific stimuli. Genetic analyses allow comparisons in the make-up and the identification of possible polymorphisms that might underlie differences in the way that individuals respond to painful stimuli and insults. Pain researchers have the challenging task to consider this wealth of knowledge regarding pain mechanisms when designing experiments, and clinicians who treat pain are anxiously waiting the time when these advances will make their treatments more effective. In one respect, the activation of the peripheral nociceptor and sensory neuron represents our first key step to our understanding of nociception. In this article, we review the key basic mechanisms associated with this phenomena and more recently identified mechanisms that are current areas of interest. Although many of these pain mechanisms apply throughout the body, we attempt to describe these mechanisms in the context of trigeminal pain.
Peripheral pain mechanisms associated with odontogenic or temporomandibular disorders and other orofacial pain conditions are generally similar to those seen elsewhere in the body. These similarities include the types of sensory neurons involved and the receptors, channels, and intracellular signaling pathways responsible for the transduction, modulation, and propagation of peripheral stimuli. Even though there are some structural features associated with the tooth pulp that make pulpal pain unique, the tooth pulp is considered as a model system to illustrate peripheral pain mechanisms associated with the trigeminal system. This also seems appropriate because toothache is a common presenting symptom for patients seeking dental care . The use of the tooth as a model system for studying pain mechanisms is well established, and advantages include a rich representation of pain fibers and that the stimulation of pulpal nerves produces mostly a pain sensation . In this regard, the tooth as a sensory organ can be considered as a specialized receptor for nociception.
The tooth pulp is composed of connective tissue that is highly vascular and rich in fibroblasts. Within this connective tissue stroma are bundles of axons that provide innervation to the tooth pulp . The distribution and overall pattern of nerve fibers within pulpal tissues have been studied extensively, including in humans and experimental animals. The majority of the axons enter the apex of the tooth, but others may enter accessory foramina when present and ascend the radicular pulp within fiber bundles composed of myelinated and unmyelinated nerve fibers ( Fig. 1 ). Nerve fibers located in these fiber tracts ascend the pulp and terminate as free nerve endings within the pulp or after entering the sub-odontoblastic plexus sequentially along this path. The sub-odontoblastic plexus is located just inside the odontoblasts and represents a fine network of many small and mostly unmyelinated fibers, many of which originate from thinly myelinated fibers. The sub-odontoblastic plexus (plexus of Raschkow) is extensive and especially elaborate in the region of pulp horns. The odontoblasts outline the entire periphery of the dental pulp and are located at the pulpodentin junction. Many of the unmyelinated nerve fibers located in the subodontoblastic plexus pass toward and terminate in the odontoblastic layer as free nerve endings, whereas others terminate in the predentin or enter dentin by way of dentinal tubules where they extend about 100 μm . Although more than 40% of dentinal tubules are innervated in the tip of pulp horns, far fewer tubules are innervated in more apical locations, with less than 1% of tubules innervated in the midradicular region . Stimulation of unmyelinated nerve fibers located in the pulp typically produces a dull throbbing and poorly localized pain sensation, whereas stimulation of the dentin produces a sharp, shooting pain that implicates the activation of more rapidly conducting myelinated fibers.
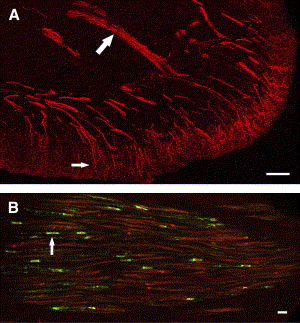
The nerve fiber density within human teeth is quite impressive. A number of ultrastructural studies have evaluated the type (as based on fiber diameter and presence or lack of myelin) and number of axons that innervate anterior and posterior teeth. Comprehensive studies of nerve fibers within posterior teeth are limited to single-rooted premolars (reviewed in ). Nair concluded that human premolar teeth contain 2300 axons at the apex; 87% of these are unmyelinated, and the remainder are myelinated. The vast majority of the myelinated fibers are thinly myelinated and fall in the A-delta class, and the remaining 7% represent the more thickly myelinated A-beta nerve fibers. Even though the “average” premolar tooth has a significant nerve density, this can vary depending on the developmental stage and type of tooth and can vary widely among individual samples. The innervation density is also dynamic because it can increase in human teeth with caries . Other axons that enter the tooth pulp originate from postganglionic sympathetic neurons located in the superior cervical ganglion and whose role involves vasoconstriction , whereas parasympathetic fibers may be lacking that provide a vasodilatory role elsewhere . Pulpal vasodilation can be achieved by the release of vasoactive neuropeptides from primary afferent terminals, a process that is integral to the production of neurogenic inflammation . This process most likely involves arterioles because these vessels are most densely innervated in the tooth pulp .
Studies in experimental animals have also described the innervation of teeth, but, unlike in human studies, these studies allow a characterization of the sensory neurons within the trigeminal ganglion that supplies the innervation to pulpal tissues. Sensory neurons that supply the tooth pulp have been identified after the retrograde transport of fluorogold to exposed dentin. These studies have found that pulpal afferents typically originate from cell bodies with small, medium, and large diameters . The cytochemistry of sensory neurons in the spinal system has been extensively evaluated, and in general these studies have identified two broad classes of neurons: (1) those that include peptidergic neurons that respond to nerve growth factor (NGF) and that express the trkA receptor and peptides such as calcitonin gene-related peptide (CGRP) and substance P (SP) and (2) nonpeptidergic neurons that respond to glial cell line–derived neurotrophic factor (GDNF) that express GDNF receptor alpha-1 and receptor tyrosine kinase (RET) and that bind the isolectin B4 (IB4). The IB4-binding neurons usually also express P2X3, an ATP-gated ion channel. In comparison, studies that have evaluated the cytochemical content of pulpal sensory neurons show some important differences when compared with the spinal system. Most notable is the lack of IB4 binding . Even though these pulpal afferents do not express IB4 binding, they do express the P2X3 receptor, which is also a marker of the nonpeptidergic class and is usually coexpressed with IB4 binding in the spinal system. A recent study has found that many of the pulpal sensory neurons express the GDNF receptor alpha-1 and that many of these coexpressed the trkA receptor . Therefore, pulpal afferent neurons express markers for peptidergic and nonpeptidergic neurons within the same neurons and do not follow patterns typically seen in the spinal system.
Many of the peptides and other molecules that have been identified as important in the activation of nociceptors in the spinal system have also been identified in the trigeminal system. Some of the peptides identified in tooth pulp include the tachykinins SP and neurokinin A , vasoactive intestinal peptide , neuropeptide Y (NPY) , methionine- and leucine-enkephalin , CGRP , cholecystokinin and somatostatin , and galanin . Peptides as a group are important in nociception because the expression of some change considerably with injury or after inflammatory insults. Sprouting of CGRP fibers is seen in the rat tooth pulp after inflammatory lesions , and similar results involving increased fibers with CGRP, NPY, SP, and vasoactive intestinal peptide have been described in human teeth with carious lesions . These same neuropeptides are especially implicated in inflammatory processes because sensory nerve stimulation can lead to their local release by way of an axon-reflex , or they may be released from nerves that innervate blood vessels, leading to vasodilation and protein extravasation, which results in a neurogenic inflammation . Neurogenic inflammation seems especially important in the trigeminal system because it represents a basic mechanism associated with the pathophysiology of migraine and may be an important event associated with the inflammation of periodontal disease and in the regulation of the immune response to infection . Neurogenic inflammation and local tissue injury are associated with the release or activation of many different molecules that are involved in the sensitization of peripheral nociceptors, including their ability to further enhance the release of CGRP and SP. These substances include cytokines, NGF, prostaglandins, histamine, bradykinin, ATP, serotonin, lipids, nitric oxide, and hydrogen ions. The local release of CGRP and SP from peripheral terminals may bind to CGRP and SP receptors on immune cells, and this binding may be involved in the regulation of the immune response in a paracrine fashion. For example, SP released from nerve terminals can bind to mast cells, leading to degranulation and the release of histamine . The degranulation of mast cells also releases tryptase, which is effective in the cleavage and activation of a new class of protease-activated receptors (PARs) and especially the PAR-2 receptor. PARs are colocalized with SP and CGRP receptors on nerve terminals and when activated can result in the additional release of SP and CGRP, thus perpetuating the inflammatory response. The effects of most neuropeptides are mediated by receptor binding, and many of these are G-protein–coupled receptors (GPCRs), which are discussed in greater detail in this article. The local release of neuropeptides that occurs in inflamed tissues represents an important event leading to the sensitization of peripheral nociceptors, and the specific mechanisms involved in this process will most likely remain a focus of pain research in the future.
Based upon these considerations, peripheral terminals of nociceptors can be envisioned as environmental detectors . Although peripheral nociceptors have a relatively simple morphology of free nerve endings ( Fig. 1 A), they are biochemically specialized by the expression and localization of various receptors and ion channels, which confer to these cells the ability to detect noxious chemical, thermal, and mechanical stimuli. These nociceptive “polymodal detectors” can trigger this local release of neuropeptides (ie, the axon reflex), leading to coordinated inflammatory and healing responses in the injured tissue and evoking action potentials that provide sensory information back to the central nervous system (CNS).
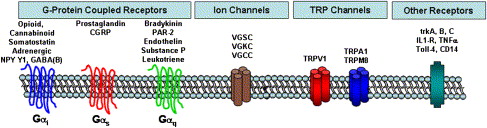
From the perspectives of understanding peripheral pain mechanisms and management, the following section reviews the major classes of receptors and ion channels that confer the ability of nociceptors to “detect” noxious changes in their peripheral area. Fig. 2 summarizes these major classes of receptors and ion channels. Understanding their pharmacology ( Table 1 ) provides insight into the pharmacologic strategies for peripheral pain control and permits appreciation of ongoing research designed to develop new peripherally acting analgesics. For example, the demonstration that dental pulp contains opioid receptors and that peripherally administered opioids reduces pain in endodontic patients suggests that locally active opioid analgesics might represent a novel class of drugs useful to treat endodontic pain patients. Because peripherally active opioid analgesics are under active development, it can be appreciated that a knowledge of peripheral pain mechanisms can improve our understanding of current and future pain control strategies .
| Receptor class | Receptor subtype | Natural ligand or stimulus | Drug | In dental pulp | In trigeminal ganglia |
|---|---|---|---|---|---|
| Opioid | Mu (MOR) | β-endorphin | Opiates | ||
| Delta (DOR) | Met-enkephalin | None available | |||
| Kappa (KOR) | Dynorphin (?) | Pentazocine | |||
| Cannabinoid | CB1 | Anandamide | None available | ||
| CB2 | ? | None available | |||
| Somatostatin | Sst1-5 | Somatostatin | Octreotide | ||
| Adrenergic | Alpha (family) | Norepinephrine | “Vasoconstrictors” | ||
| Beta (family) | Epinephrine | Albuterol, etc. | |||
| GABA | GABA (B) | GABA | Baclofen | ||
| NPY | Y1 (etc.) | Neuropeptide Y | None available | ||
| Prostaglandin | EP2, EP3 (etc.) | PGE2 | NSAIDs, steroids | ||
| CGRP | CGRP-R1, -R2 | CGRP | CGRP28-37 | ||
| Bradykinin | B1 | Kallidin | Des-Tyr HOE140 | ||
| B2 | Bradykinin | HOE140, steroids | |||
| Protease (PAR) | PAR2 (etc.) | ? | |||
| Endothelin | ETA, ETB | Endothelin | Atrasentan (antagonist) | ? | |
| Substance P | NK1, NK2 | Substance P | L-703,606 | ||
| Leukotriene | BLT1, 2 and CysLT1, 2 | LTB4, LTD4 | Monteleukast, zafirlukast | ||
| VGSC | Na v 1.1 (etc.) | Local anesthetics | |||
| VGCC | ? | ||||
| VGKC | ? | ||||
| TRP | TRPV1 | Heat, acid, some lipid-like compounds | Capsaicin | ||
| TRPA1 | Cold (?) | Mustard oil, garlic | |||
| TRPM8. | Cold | Menthol | |||
| Trk | trkA (etc.) | ? | |||
| Cytokine | IL1-RI, IL1-RII | IL1 | Anakinra | ||
| TNF | TNFα | Adalimumab | |||
| Innate PAMPS | Toll4 | Endotoxin (LPS) | |||
| CD14 | Endotoxin (LPS) |
Mechanisms for detecting stimuli and clinical implications
G-protein–coupled receptors
The G-protein–coupled receptors (GPCRs) comprise a large superfamily of receptors. The GPCRs share a common structure (seven transmembrane regions on the protein) and are called “G-protein–coupled” because they share a common signaling mechanism via activation of a certain class of GTP-binding proteins (aka G-proteins). Thus, the GPCR undergoes a conformational change when a drug or endogenous substance binds to the receptor, resulting in the GPCR binding to a G-protein and initiating a second messenger signaling pathway . Although there are many subtypes of G-proteins and second messenger systems, and the actual signaling pathways are far more complicated than space permits, for our purposes we focus on the three major subtypes of G-proteins: Gα i/o , Gα s , and Gα q and their classic signaling pathways.
GPCRs that are coupled to the Gα i/o signaling pathway include opioid, cannabinoid, somatostatin, certain adrenergic subtypes, NPY, and GABA(B) receptors. In general, activating a Gα i signaling pathway leads to the inhibition of neuronal function by reducing cAMP levels, opening certain potassium channels (leading to a more negative membrane potential, called “hyperpolarization,” and thus reducing the probability of triggering an action potential) and inhibiting certain calcium channels. As a first approximation, drugs that activate the Gα i GPCRs that are expressed on nociceptors would be predicted to be peripherally active analgesics. Drugs that activate peripheral opioid, cannabinoid, adrenergic, Y1, or GABA(B) receptors produce peripheral analgesia or inhibit peripheral neuronal function . Clinicians use several drugs that activate Gα i GPCRs, and many additional drugs are in development as analgesics that act by these mechanisms.
In many respects, the Gα s GPCRs are complimentary to the Gα i family of GPCRs because these receptors typically increase cAMP levels, leading to cellular excitation. Examples of GPCRs that are coupled to the Gα s signaling pathway include prostaglandins and CGRP ( Table 1 ). Recent molecular studies have demonstrated that of the four known subtypes of prostaglandin receptor, only the EP2 and EP3 subtypes are expressed in trigeminal sensory neurons . Thus, local increases in prostaglandin E2 in dental pulp or periradicular exudates are likely to contribute to odontogenic pain mechanisms via activation of EP2 or EP3 receptors expressed on trigeminal sensory neurons. Although EP receptor antagonists have been developed, the current clinical strategy to control this receptor system is via the use of nonsteroidal anti-inflammatory drugs (NSAIDs) or via glucocorticoid steroids. Both classes of drugs block prostaglandin synthesis by interfering with the function of cyclooxygenase I/II (NSAIDs) or with phospholipase A2 (steroids).
Several GPCRs are coupled to the Gα q signaling pathway, including bradykinin, protease-activated receptors, endothelin, SP, and leukotriene receptors. In general, activation of a Gα q –coupled GPCR leads to activation of the phospholipase C/protein kinase C signaling pathways. This can evoke a considerable stimulatory effect on nociceptors, leading to sensitization of the capsaicin receptor, transient receptor potential V1(TRPV1). Recent studies have demonstrated that activation of the phospholipase C signaling pathway can reduce the normally high threshold for activating TRPV1 from temperatures of ∼43°C to as low as ∼37°C . This would lead to spontaneous activation of TRPV1 at body temperatures, possibly contributing to the spontaneous pain in patients who have irreversible pulpitis or acute apical periodontitis or other orofacial pain conditions. Prior studies have provided evidence for activation or functional activity of the bradykinin, endothelin, SP, and leukotriene systems in dental pulp .
Voltage-gated ion channels
Voltage-gated ion channels (VGICs) are transmembrane, pore-forming proteins that allow the selective passage of certain ions in a voltage-dependent manner. There are more than 140 members of this superfamily representing one of the largest collections of proteins involved in signal transduction . They also represent key therapeutic targets given their importance in transduction. Within this superfamily are several important classes of ion channels that include the potassium (K + ), calcium (Ca 2+ ), and sodium (Na + ) VGICs. The activation of these classic channels is a key process involved in the initiation and propagation of action potentials and in the release of neurotransmitters involved in synaptic transmission. Their importance in pain pharmacology is recognized because analgesics exist that function directly on the Na + and Ca 2+ VGICs, and the actions of many different drugs produce analgesia indirectly through effects on K + channels.
There is a great deal of similarity in the structure of these different classic VGICs, and this homology suggests a similar origin of not only these classic channels but of the entire superfamily . The K + channels represent the ones with the simplest structure, whereas the Ca 2+ and Na + channels represent modifications of this structural motif. The Na + channel was the first of these to be described and consists of an alpha subunit consisting of four homologous domains (I–IV) that surround a central pore for ion passage . In addition to the pore, the alpha subunit contains a selectivity filter that allows only certain types of ions to pass and a voltage-sensor that allows a conformational change and opening of the pore based on voltage. Each domain consists of six transmembrane α-helices referred to as S1 through S6. The structure of the Ca 2+ channel is similar to Na + channels , whereas the K + channel consists of a tetramer of an identical protein monomer that resembles one homologous domain of Na + and K + channels . Auxillary subunits are typically associated with the α-subunit, and, in the case of Na + channels, these beta subunits can modulate the expression, localization, and gating properties of the α-subunits and thus represent possible therapeutic targets. Summary statements regarding the distributions, functional significance, and possible therapeutic roles of each of the channels included in the superfamily of voltage-gated ion channels have recently been published. These statements include descriptions of the standardized nomenclature used to denote the different members of this class .
Although it is difficult and most likely unfair to summarize the contribution of each of these classic VGICs in neuronal function, the following generalizations can be made. The activation of Na + channels is critical for action potential (nerve impulse) initiation and propagation. The opening of the voltage-gated Na + channels occurs when a transient generator potential is created by the activity of other ion channels (such as transient receptor potential [TRP]), thus reaching the critical level needed to open the pore. If enough Na + ions enter the axon, a depolarizing threshold is reached, resulting in the initiation of an action potential. Thus, drugs that block sodium channels (eg, lidocaine) play a critical role in dental therapeutics. The activation of the K + channel is necessary to hyperpolarize (bringing the resting potential within the nerve membrane back to a negative potential) and thus terminating the action potential. Therefore, the activation of the classic voltage-gated Na + channel initiates this activity, whereas the opening of K + channel results in the termination of nerve activity. The role of the voltage-gated Ca 2+ channels in nerve activity is more complex because calcium entry into neurons can produce profound short- and long-lasting effects on many different cellular functions due to its role as a second messenger and involvement in intracellular signaling pathways. Important functions of voltage-gated Ca 2+ channels include its influence on cell body excitability and the ability to gate the entry of calcium into nerve terminals, leading to vesicle fusion and release of neurotransmitter during synaptic transmission. Each of these channels, and especially the various subtypes, represents possible therapeutic targets to control the altered excitability of nociceptors. Evidence for the role of each of these classic VGICs and related members in pain conditions is discussed below.
Sodium channels: the Na v s
Much recent interest has been focused on the contribution of altered voltage-gated sodium channel expression to pain states . The importance of sodium channels on pain transmission is well known because the successful practice of “painless” dentistry largely depends on the sodium channel blocking effect of local anesthetics. Sodium channels are important in action potential initiation and propagation in response to normal stimuli , but they also seem to have a role in increased neuronal excitability and especially spontaneous and ectopic activity associated with inflammatory and neuropathic pain states. The association of altered sodium channel function with basic neuropathic pain mechanisms is strengthened by the relative effectiveness of medications with a sodium channel blocking effect, such as the anticonvulsant carbamazepine in the treatment of neuropathic pain conditions and especially trigeminal neuralgia . The tricyclic antidepressants also represent a useful neuropathic pain medication, and some of their effectiveness may be due to a sodium channel–blocking effect .
Sodium channels are recognized as a diverse group consisting of at least nine different subtypes, or isoforms, localized to nervous system tissues and designated as Na v 1.1 through 1.9 . Although all nine show similarities in structure and as a group show more similarity in function than the Ca 2+ and K + families, some important differences exist. These include a differential nervous system distribution and important differences in expression after inflammatory or axotomy insults . The relative differences in expressions are important physiologically because each sodium channel has unique gating properties that can influence action potential initiation. The isoforms that are normally expressed in sensory neurons include the Na v 1.1, -1.2, -1.6, -1.7, -1.8, and -1.9 isoforms. The Na v 1.1, -1.2, and -1.6 isoforms are also found in the CNS, whereas Na v 1.3 is seen in the developing nervous system . The Na v 1.6 isoform is the predominant sodium channel located at nodes of Ranvier throughout the nervous system and thus is critically linked to the saltatory conduction of action potentials in myelinated fibers. The Na v 1.7, -1.8, and -1.9 isoforms are preferentially expressed in the peripheral nervous system and seen in a subset of nociceptors . Their peripheral nervous system location makes them attractive targets for the development of pharmacologic agents because such agents may lack the CNS side effects associated with many of the current medications that block sodium channels, such as anticonvulsants.
Nerve injury models have implicated the Na v 1.3, -1.7, -1.8, and -1.9 isoforms in the generation of neuropathic pain. Nerve injury models result in an upregulation of the previously non-expressed Na v 1.3 gene in DRG neurons , in dorsal horn neurons , and in dorsal horn and thalamic neurons after spinal cord injury . Peripheral nerve injury is associated with a downregulation or loss of Na v 1.8 and Na v 1.9 in the DRG but with fewer changes of both isoforms at the site of injury . The Na v 1.8 isoform has also been implicated in nerve injury hyperalgesia , including an upregulation in nearby uninjured c-fibers . Axotomy of the inferior alveolar nerve also decreases Na v 1.8 mRNA in trigeminal ganglion neurons, like most studies done in the spinal system . Recent human studies have also found increased Na v 1.7, -1.8, and -1.9 immunoreactivity and protein in injured nerves, an association of increased Na v 1.8 with hyperalgesia, and decreased expression in the injured DRG neurons . Primary erythermalgia, a disease characterized by sporadic attacks of swollen, red, and warm extremities, has recently been defined as a neuropathic pain disorder due to mutations in the SCN9A gene that encodes for the Na v 1.7 protein . Other recent findings show no change in neuropathic pain behavior in rats treated with Na v 1.3 antisense oligonucleotides and in knockout mice lacking Na v 1.7 and -1.8 , whereas a specific blocker of Na v 1.7 and -1.8 and Na v 1.8 inhibited neuropathic behaviors. The role of altered sodium channel expression in neuropathic pain states remains an active area of research. The recent development of isoform-specific blockers is encouraging, and the development of other specific blockers should help to define the role of altered isoform expression to the development of neuropathic pain states.
Other studies have evaluated the effect of inflammation on specific isoform expression, and these results have suggested the involvement of Na v 1.7 and the tetrodotoxin-resistant isoforms Na v 1.8 and -1.9 in inflammatory pain mechanisms . The expression of these isoforms may be mediated through prostaglandin signaling , and pretreatment with ibuprofen can prevent the augmentation of Na v 1.7 and -1.8 seen after injection of complete Freud’s adjuvant . Nerve growth factor is also involved in the expression of Na v 1.7 and Na v 1.8 . There is interest in studying sodium channel expression in human dental pulp , and recent studies have shown an increase in Na v 1.8 in painful tooth pulp when compared with normal tooth pulp . One possible consequence of an increased expression is a higher incidence of local anesthesia failures encountered when treating painful teeth . Although differences in the expression of the various isoforms are seen after nerve injury and inflammatory insults, the isoform that contributes most to the development of altered neuronal excitability is unknown.
Potassium channels: the voltage-gated potassium channels and others
The potassium-selective channels represent the largest class of ion channels and consist of diverse subtypes. The voltage-gated potassium (K v ) channels are one subtype and represent about 40 of the 70 known potassium-selective channels. Other K + –selective channels include the inward rectifying, two-pore, and Ca 2+ –activated K + channels. The Ca 2+ –activated K + channels include the big, intermediate, and small conductance K + channels.
Each of the K v genes encodes a single peptide subunit. The active K v channel is composed of four subunits that can be homotetramers of the same subunit or heterotetramers composed of various subunits from within the family. The K v family members, as designated with the IUPHAR nomenclature and followed by the HUGO Gene Nomenclature Committee nomenclature in parentheses, include K v 1.1–1.8 (KCNA1–7, 10), K v 2.1–2.2 (KCNB1–2), K v 3.1–3.4 (KCNC1–4), K v 4.1–4.3 (KCND1–3), K v 5.1 (KCNF1), K v 6.1–6.4 (KCNG1–4), K v 7.1–7.5 (KCNQ1–5), K v 8.1–8.2 (KCNV1–2), K v 9.1–9.3 (KCNS1–3), K v 10.1–10.2 (KCNH1–2), K v 11.1–11.3 (KCNH2,6,7), and K v 12.1–12.3 (KCNH8,3,4). The K v 7 family represents the most interesting family from a pharmacologic aspect because mutations in four of the subunits have been associated with diseases such as long QT syndrome, deafness, and seizures. The K v 7.2 to 7.5 subtypes are considered possible targets for the development of anticonvulsants, and, due to the effectiveness of other anticonvulsants in neuropathic pain management, they may also represent pharmacologic targets for pain management. This association seems to hold true because the anticonvulsant retigabine (an opener of the K v 7.2–7.5 subtypes) seems effective in some models of neuropathic and chronic pain . Other K v subtypes that may be implicated in pain include K v 1.4, which is found in small-diameter dorsal root ganglion neurons , and the K v 4.2 subtype, which is localized to dorsal horn neurons and when inactivated by extracellular signal-related kinase after injury is inactivated and no longer able to inhibit neuronal firing .
Other K + channels that are implicated in pain mechanisms include two members of the inward rectifying family, the K ATP subtype and the G-protein regulated inward rectifier K + channels (GIRK or K ir 3). The K ATP subtype is implicated in peripheral analgesia because peripheral injections of the specific blockers pinacidil and diazoxide produced antinociception in a paw pressure test . The opening of K ATP and GIRK channels seems to be a critical step in the antinociceptive effects of many analgesic medications, including opioids. This occurs indirectly because opioid binding activates G i/o proteins, which open the K ATP , GIRK1, and GIRK2 subtype K + channels, thus contributing to antinociception . Opioid receptors located spinally primarily affect GIRK1 and GIRK2 , whereas peripheral opioid analgesia primarily affects the K ATP K + channels . The activation of GPCRs by non-opioid agonists and the subsequent opening of K + channels underlie the analgesic action of many different medications, including adrenoceptors, adenosine, 5-HT1A receptor agonists, muscarinic and dopamine receptors, cannabinoid receptors, GABA B receptors, some NSAIDs, tricyclic antidepressants, antihistamines, and gabapentin . Evidence suggests the involvement of the big and small conductance K + channel subtypes of the Ca 2+ –activated K + channel family in some of these effects. In summary, the activation and subsequent opening of a number of different K + channels seems to be a promising area of research that may lead to the development of new classes of analgesics through a direct opening effect on K + channels or indirectly through the activation of GPCRs.
Calcium channels: the voltage-gated Ca 2+ channels and a few others
Activation of the voltage-gated Ca 2+ (Ca v ) channels have broad-reaching effect on cellular function due to the role of calcium as an important intracellular second messenger system in addition to critical roles in the control of neuronal excitability and the release of neurotransmitters. The structure of the Ca v is similar to that of the Na v , consisting of four homologous domains with each domain consisting of a six-transmembrane α-helix segment . The α1 subunit may also be associated with β and α2-δ and -γ subunits, which modify the gating characteristics of the α1 subunit. Currents due to calcium channel activation were initially characterized based on their physiologic properties (L, N P/Q, and R) and then by an alphabetical nomenclature based on that used to classify the K v . This classification includes Ca v 1.1 through Ca v 1.4 (L current), Ca v 2.1 (P/Q current), Ca v 2.2 (N current), Ca v 2.3 (R current), and Ca v 3.1 through Ca v 3.3 (T current).
The Ca v 2 (P/Q, N, and R currents) channels are of great interest with regard to pain mechanisms because they are blocked by peptides isolated from the venom of spiders and snails . Ziconotide represents a new intrathecally administered analgesic that is approved for chronic pain resistant to other therapies and that specifically blocks activity at the Ca v 2.2 channel and its N current . It is a synthetic peptide based on the venom from the marine snail Conus magus and inhibits the release of excitatory amino acids from primary afferent terminals . Inhibition of the N current is also achieved with the opioid receptor like receptor 1 agonist, nociceptin, which triggers a PKC-dependent internalization of N-type channels . Mice lacking the Ca v 2.2 channel show decreased inflammatory and neuropathic pain behaviors , suggesting important influences of this channel on the neurotransmission of nociceptive stimuli. The analgesic action of various venoms may not be limited to the Ca v 2.2 channel because the venom from the spider Phoneutria nigriventer inhibits all three currents associated with the Ca v 2 channels . The Ca v 3.2, T-type channel also represents a target because antisense knockdown of this gene inhibits pain behaviors in a neuropathic pain model . Other potential targets related to the Ca v include the β3 and the α2-δ type 1 subunits. Pregabalin is another new medication that is related to gabapentin. Pregabalin is approved to treat postherpetic neuralgia and diabetic neuropathy pain, and its analgesic action may include its ability to bind to the α2-δ type 1 subunit . The modification of Ca 2+ currents by drugs and venoms has led to the development of new analgesics, and hopefully more medications with specific action and fewer side-effects will be developed in this class.
The transient receptor potential channels
The TRP channels represent a family of six different members including some that that act broadly in the transduction of sensory stimuli related to pain, temperature, vision, hearing, taste, and pheromone detection . Most are weakly gated by voltage and as a class act as nonselective cation channels that allow the passage of Na + , sometimes Mg 2+ , and especially Ca 2+ into cells. Because Ca 2+ plays an important role as an intracellular second messenger, they are implicated in the control of many cellular processes, including exocytosis, contraction, apoptosis, migration, cell development, and neuronal excitability. They often work in concert with other receptors, including GCPRs and tyrosine kinases. Tyrosine kinase activates phospholipase C, leading to Ca 2+ release from the endoplasmic reticulum . The TRP family is somewhat related in structure to the K + channels and consists of six transmembrane loops. They can form homomeric functional units or can form associations with other members, allowing the formation of heteromeric units. The six subfamilies of the TRPs include the vanilloid receptor TRPs (TRPVs), the melastatin or long TRPs (TRPMs), the ankyrin transmembrane protein 1 (ANKTM1 or TRPA1), the classic TRPs, the mucolipins, and the polycystins . Four individual members within these subfamilies (TRPV1, TRPV2, TRPM8, and TRPA1) have been strongly implicated in pain signaling or some aspects of thermoreception, and all allow the passage of Ca 2+ preferentially more than other cations.
The TRPV subfamily consists of six different members (TRPV1–6); TRPV1 is the most intensely studied and best understood member of this group . The TRPV1 receptor was the first molecule to be found that is gated by temperature (≥ 43°C) and that represents the capsaicin receptor and was first described as the vanilloid receptor (VR1). It is primarily a Ca 2+ –permeable channel that is also gated by hydrogen ions and polyunsaturated fatty acids and represents a possible receptor for the endogenous cannabinoid anandamide . TRPV1 knockout mice have shown that the expression of this ligand-gated channel is critical for the development of inflammatory hyperalgesia but is not necessary for the detection of heat in normal (uninflamed) tissues . Capsaicin produces a neurogenic inflammation (by evoking peripheral neuropeptide release) and primary and secondary hyperalgesia and highlights the importance of this receptor in this process. The receptor can be sensitized by inflammatory mediators such as bradykinin, prostaglandins, serotonin, ATP, and adenosine . This sensitization can lead to a potentiation of currents through the channel and can result in lowering the temperatures needed to activate the channel . The TRPV1 channel also shows the ability to become desensitized after prolonged stimulation. The TRPV1 channel represents an attractive pharmacologic target because it is widely expressed in the trigeminal and dorsal root ganglion in small-diameter cell bodies that typically give rise to unmyelinated axons, consistent with a nociceptive phenotype . The TRPV1 receptor is seen in nociceptive tissues such as the human tooth pulp , is found in peptidergic and nonpeptidergic sensory neurons , and may play a role in neuropathic pain mechanisms . Given the importance that TRPV1 plays in inflammatory pain mechanisms, it is widely regarded as a phenotypic marker for nociceptive neurons.
The TRPV2 receptor is another member of the TRPV subfamily and may play a role in the detection of noxious heat . It is activated by higher temperatures, with a threshold of 52°C, and is not activated by protons or the vanilloids. The TRPV2 receptor is seen in larger cell bodies within sensory ganglia, A-delta and A-beta myelinated fibers, and in different cells than TRPV1 . More recently, the heat receptor story has been complicated by the finding that TRPV1 and TRPV2 knockout mice respond normally to heat responses . This surprising finding has led to their hypotheses that under normal conditions there is a population of IB4-negative neurons that respond to heat in ways that do not involve TRPV1 and TRPV2 and that TRPV1 is active only after injury or in disease states. Even though TRPV1 and TRPV2 have been implicated in the processing of noxious heat stimuli, additional studies are needed to more fully describe the role of each in this response.
The TRPM8 receptor was first called the cold menthol-receptor 1 because it binds menthol, thus producing its cooling effect . Activity is increasingly activated by cool and noxious cold temperatures that range from 28 to 8°C and by other cooling compounds, such as icilin . The activation by menthol is important because this compound can initiate a painful response when applied to human skin , leading to cold allodynia but without the development of an axon reflex . Menthol can also produce a sensitization of the receptor, leading to an increase in the temperature, which is needed to activate the channel. TRPM8 has been identified in primary afferent neuronal cell bodies that give rise to myelinated A-fibers and unmyelinated C-fibers and has been seen in a different subset of neurons than TRPV1 . This study also classified the different TRP receptors based on their associations with trk receptors and found an important correlation of TRPM8 with the high-affinity nerve growth factor receptor trkA, which is seen as a marker of neurons that are critically involved in inflammatory pain mechanisms . These studies have identified the TRPM8 receptor as a cold receptor.
The TRPA1 receptor (first called ANKTM1) was initially identified as a cold receptor . It is activated by temperatures below 17°C, but conflicting evidence exists regarding its role in the signaling of noxious cold . This receptor is activated by different naturally occurring compounds (eg, allyl isothiocyanates, such as mustard oil and wasabi; thiosulfinate allicin, which is found in garlic; and cinnamon) that, when applied to skin, can produce pain and neurogenic inflammation. Activation of TRPA1 also mediates the inflammatory response to environmental irritants, such as tear gas and car exhaust , bradykinin , and some of the effects of peripherally administered cannabinoids . This receptor may also be involved in the detection of mechanical stimuli , an ability that may be related to the presence of multiple ankyrin repeats also seen in the TRPN1 receptor that mediates mechanotransduction in flies . The TRPA1 receptor is found mainly in a subset of unmyelinated nociceptors and is colocalized in a subpopulation of TRPV1 neurons . Due to its role in inflammation, activation by many different compounds that produce pain when applied topically, and its colocalization with TRPV1, it represents an important receptor involved in the transduction and modulation of painful stimuli.
In summary, the TRPs represent an important class of receptors involved in the pain associated with peripheral inflammation. The activation of some of these receptors by thermal stimuli in pathologic conditions represents an important finding. Given the ability of thermal stimuli and especially cold in the production of a painful response in inflamed teeth, the evaluation of these receptors in pulp from normal and painful human extracted teeth could provide additional insights into thermally mediated pain mechanisms.
Several other receptor systems are expressed on nociceptors and modulate the activity of this important class of sensory neurons ( Table 1 ). The neurotrophin receptors trkA, trkB, and trkC are expressed on sensory neurons and detect tissue levels of NGF, BDNF, and GDNF, respectively. Of particular interest to dentists, NGF has been shown to increase during pulpal inflammation , to sensitize TRPV1 , and to evoke hyperalgesia after injection in human volunteers . Inflammation likely increases more than one neurotrophin, and these potent mediators significantly alter sprouting of trigeminal neurons .
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


