Allogeneic hematopoietic cell transplantation (allo-HCT) is used for the treatment of a variety of disorders, primarily hematologic malignancies. Graft-versus-host disease (GVHD) is a significant complication following allo-HCT and a major cause of morbidity and mortality. The oral cavity is frequently involved in GVHD, leading to pain, functional impairment, and reduced quality of life. Early diagnosis, management, and long-term follow-up of oral GVHD are important components of overall patient care.
Key points
- •
Allogeneic hematopoietic cell transplantation (allo-HCT) is used for the treatment of a variety of disorders, primarily hematologic malignancies.
- •
Graft-versus-host disease (GVHD) is a significant complication following allo-HCT and a major cause of morbidity and mortality.
- •
The oral cavity is frequently involved in GVHD, leading to pain, functional impairment, and reduced quality of life.
- •
Early diagnosis, management, and long-term follow-up of oral GVHD are important components of overall patient care.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is considered to be a curative treatment of many hematologic malignancies as well as for a wide range of hematologic and immune deficiency states and immune diseases. Graft-versus-host disease (GVHD, Box 1 ), an immunologically mediated disease, is a major cause of morbidity and mortality after allo-HCT and the most significant barrier to treatment success.
A clinical syndrome that results from an immunologic attack by donor immunocompetent T cells on patient tissues, directly or through exaggerated inflammatory responses, following allogeneic HCT
In 1966, Billingham outlined the 3 fundamental requirements for GVHD. First, the graft must contain immunologically competent cells; second, the host must express tissue antigens that seem foreign to the graft; and finally, the host must be incapable of rejecting the donor graft. As stipulated in the third precondition, recipients undergo myelosuppressive and immunosuppressive regimens before allo-HCT, reducing the risk of graft rejection. After transplantation, donor-derived immunocompetent T lymphocytes may react against histocompatibility antigens on the recipient cells and induce immune responses, resulting in recipient tissue damage.
The major risk factor for GVHD development is human leukocyte antigens (HLA) disparity. HLAs are expressed on the cell surfaces of all nucleated cells in the body and are encoded by the major histocompatibility complex (MHC) located on the short arm of chromosome 6. If incompatible with the recipient, the donor’s immune cells mount an alloimmune response against antigens on the recipient cells leading to GVHD. Careful matching of donor and recipient HLA reduces the risk for GVHD (as in sibling HLA-matched grafts). The current guidelines recommend high-resolution DNA-matching (ie, indicating that the donor and recipient express identical alleles) for the following 4 HLA loci in order to maximize post-HCT survival: HLA-A, -B, -C, and -DRB1 (8/8 match). Ideally, unrelated donors should be genotypically identical to the recipients at all HLA loci; however, finding the ideal match is often extremely challenging in light of the millions of potential haplotypes existing. Furthermore, despite selection of an HLA-matched donor, T lymphocyte reaction may still be elicited by minor histocompatibility antigens encoded by non-MHC loci, contributing to GVHD development. Other important factors determining GVHD development and severity are listed in Box 2 .
- •
Donor/recipient sex mismatch (especially female donors to male recipients)
- •
Increasing recipient age
- •
Choice of progenitor cells source
- •
Pretransplantation manipulations and graft composition
- •
Intensity of conditioning regimen
Along with the increase in morbidity and mortality, GVHD is also associated with a reduced incidence of malignancy relapse due to the graft-versus-tumor (GVT) effect. It is thought that this is mediated by donor T lymphocytes that recognize tumor antigens and elicit antitumor responses. Successful transplantation results from carefully balancing GVT effects against chronic GVHD (cGVHD). Overtreatment of cGVHD may adversely impact malignancy relapse rates, whereas too robust a GVT effect, such as that achieved by donor lymphocyte infusion (DLI) for patients who relapse after HCT, may induce severe GVHD with its attendant morbidities.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is considered to be a curative treatment of many hematologic malignancies as well as for a wide range of hematologic and immune deficiency states and immune diseases. Graft-versus-host disease (GVHD, Box 1 ), an immunologically mediated disease, is a major cause of morbidity and mortality after allo-HCT and the most significant barrier to treatment success.
A clinical syndrome that results from an immunologic attack by donor immunocompetent T cells on patient tissues, directly or through exaggerated inflammatory responses, following allogeneic HCT
In 1966, Billingham outlined the 3 fundamental requirements for GVHD. First, the graft must contain immunologically competent cells; second, the host must express tissue antigens that seem foreign to the graft; and finally, the host must be incapable of rejecting the donor graft. As stipulated in the third precondition, recipients undergo myelosuppressive and immunosuppressive regimens before allo-HCT, reducing the risk of graft rejection. After transplantation, donor-derived immunocompetent T lymphocytes may react against histocompatibility antigens on the recipient cells and induce immune responses, resulting in recipient tissue damage.
The major risk factor for GVHD development is human leukocyte antigens (HLA) disparity. HLAs are expressed on the cell surfaces of all nucleated cells in the body and are encoded by the major histocompatibility complex (MHC) located on the short arm of chromosome 6. If incompatible with the recipient, the donor’s immune cells mount an alloimmune response against antigens on the recipient cells leading to GVHD. Careful matching of donor and recipient HLA reduces the risk for GVHD (as in sibling HLA-matched grafts). The current guidelines recommend high-resolution DNA-matching (ie, indicating that the donor and recipient express identical alleles) for the following 4 HLA loci in order to maximize post-HCT survival: HLA-A, -B, -C, and -DRB1 (8/8 match). Ideally, unrelated donors should be genotypically identical to the recipients at all HLA loci; however, finding the ideal match is often extremely challenging in light of the millions of potential haplotypes existing. Furthermore, despite selection of an HLA-matched donor, T lymphocyte reaction may still be elicited by minor histocompatibility antigens encoded by non-MHC loci, contributing to GVHD development. Other important factors determining GVHD development and severity are listed in Box 2 .
- •
Donor/recipient sex mismatch (especially female donors to male recipients)
- •
Increasing recipient age
- •
Choice of progenitor cells source
- •
Pretransplantation manipulations and graft composition
- •
Intensity of conditioning regimen
Along with the increase in morbidity and mortality, GVHD is also associated with a reduced incidence of malignancy relapse due to the graft-versus-tumor (GVT) effect. It is thought that this is mediated by donor T lymphocytes that recognize tumor antigens and elicit antitumor responses. Successful transplantation results from carefully balancing GVT effects against chronic GVHD (cGVHD). Overtreatment of cGVHD may adversely impact malignancy relapse rates, whereas too robust a GVT effect, such as that achieved by donor lymphocyte infusion (DLI) for patients who relapse after HCT, may induce severe GVHD with its attendant morbidities.
Pathobiology and incidence of GVHD
Traditionally, GVHD has been classified as either acute (aGVHD) or chronic according to the time of clinical onset after transplantation. Any GVHD manifestations before day +100 after transplantation were defined as aGVHD, and any manifestations present after day +100 were defined as cGVHD. Recently, however, it has been accepted that acute and chronic GVHD represent a clinical continuum with distinct clinical features and underlying pathophysiologic mechanisms. aGVHD is characterized by strong inflammatory features, whereas cGVHD displays more autoimmune and fibrotic characteristics ( Table 1 ). The current consensus, therefore, is that aGVHD and cGVHD should be differentiated according to clinical manifestations and pathologic features rather than merely the time of onset after transplantation. The National Institutes of Health (NIH) Consensus Development Project on Criteria for Clinical Trials in cGVHD has further defined 2 subcategories for both aGVHD and cGVHD. Acute GVHD can be classic, occurring within 100 days after HCT, or persistent, recurrent, or late when occurring beyond day +100 after HCT (for example, in cases of nonmyeloablative conditioning). Similarly, cGVHD can be either classic (ie, without concomitant features or characteristics of aGVHD) or can manifest in an overlap syndrome with features of both cGVHD and aGVHD occurring simultaneously as can be seen in patients receiving DLI.
| Organ or Site | aGVHD a | cGVHD b |
|---|---|---|
| Skin | Erythema Maculopapular rash Pruritus |
Poikiloderma Lichen planus–like features Sclerotic and morphealike features |
| Oral | Gingivitis Mucositis Erythema Pain |
Lichen planus features Hyperkeratotic plaques Restriction of mouth opening from sclerosis |
| GI tract | Anorexia Nausea Vomiting Diarrhea Weight loss |
Esophageal web Strictures or stenosis in the upper to mid third of the esophagus |
| Genitalia | Lichen planus–like features Vaginal scarring or stenosis |
|
| Liver | Total bilirubin, alkaline phosphatase >2 times upper limit of normal ALT or AST >2 times upper limit of normal |
|
| Lung | Bronchiolitis obliterans | |
| Muscles, joints | Fasciitis Joint stiffness or contractures secondary to sclerosis |
a Manifestations that can also be found in cGVHD.
b Diagnostic features. These features are sufficient to establish the diagnosis of cGVHD.
aGVHD
The pathobiology of aGVHD is considered to be a 3-step process, starting with tissue damage induced by the pretransplantation conditioning regimen, followed by donor-derived T lymphocyte activation and clonal expansion, and ultimately tissue destruction induced by immune and inflammatory responses in the effector step. In the first step, the conditioning regimen, comprised of chemotherapy, radiation therapy, and/or immunosuppressive medications, induces tissue damage and subsequent activation of patient antigen presenting cells (APCs). In the second step, APCs present the patients’ alloantigens to donor-derived T lymphocytes, leading to their activation characterized by a primarily T-helper 1 (Th1) response. In step 3, the effector T lymphocytes, along with natural killer cells, macrophages, and proinflammatory cytokines, inflict direct tissue damage, as well as indirect damage, through intense inflammatory responses associated with the massive cytokine production, known as the cytokine storm .
The incidence of aGVHD varies with relation to several immunologically based factors, primarily the degree of donor-recipient histocompatibility ( Table 2 ). The primary target organs of aGVHD are the skin, the liver, and the gastrointestinal (GI) tract. Other organs may also be involved, including the oral cavity. The severity of overall aGVHD is graded from I to IV by combining each of the 3 target organs’ clinical stage of involvement. The grade of aGVHD has long been known to correlate with overall survival, with the best survival for patients with grade I disease. This correlation was demonstrated by the Chronic Leukemia Working Party of the European Group for Bone Marrow Transplantation, who reported transplant-related mortality rates for grades I to IV aGVHD to be 27%, 43%, 68%, and 92%, respectively, in 1294 patients after allo-HCT for chronic myeloid leukemia.
| Matched (%) | Mismatched (%) | |
|---|---|---|
| Related | 20–50 | 75–80 |
| Unrelated | 70 | 80–90 |
cGVHD
The mechanisms underlying cGVHD are not well understood. Two basic theories for the pathobiology of cGVHD have been proposed. According to the first theory, cGVHD is simply an end-stage alloreactivity in which donor T lymphocytes have differentiated toward a Th2 phenotype, associated with B-cell activation and autoantibody production. The second theory suggests that cGVHD is the outcome of poor/dysfunctional immunologic recovery after HCT, involving altered peripheral mechanisms of tolerance and decreased negative selection related to impaired thymic function, resulting in an increase in peripheral autoreactive T lymphocytes that generate further activation of effector functions (cytokine secretion, cytolytic activity, and antibody production). Although the association of cGVHD with altered B-cell homeostasis is well established, the exact role of B lymphocytes in the pathogenesis of GVHD remains unknown. In the clinical setting, reduction of alloreactive B lymphocytes responses using rituximab, a CD20-specific monoclonal antibody, can be effective in the treatment of cGVHD.
Three patterns of cGVHD onset have been identified: (1) de novo onset, without prior aGVHD; (2) quiescent onset, following a recovery period without any GVHD manifestations (postresolution of aGVHD); and (3) progressive onset, with cGVHD directly following aGVHD. The manifestations of cGVHD may be limited to one organ or widespread and most frequently involve the skin, eyes, oral cavity, GI tract, liver, and lungs. With reported incidence rates ranging from 25% to 80% in long-term survivors, cGVHD is the most common late complication following allo-HCT and the leading cause of nonrelapse mortality. The incidence and severity of cGVHD are correlated with multiple factors, including HLA disparity, increasing recipient age, sex mismatch, and source of progenitor cells (see Box 2 ). However, the most important risk factor for cGVHD development is the diagnosis of previous aGVHD.
Oral GVHD
Incidence
Although not a classic target organ of aGVHD, oral manifestations may be encountered, clinically presenting as mucosal erythema, ulcerations, and painful desquamative lesions. Because these manifestations are nonspecific, oral aGVHD may be mistaken for or obscured by several other conditions, including conditioning-induced mucositis and herpes simplex virus (HSV) infection. However, because aGVHD typically develops well after engraftment and resolution of mucositis, and acyclovir prophylaxis is highly effective in preventing reactivation of HSV, these entities can be readily differentiated. The incidence of oral aGVHD is unknown but is thought to be exceedingly rare. In contrast, the oral cavity is one of the most frequently affected sites in cGVHD, with more than 70% of patients who develop cGVHD demonstrating oral involvement.
Clinical Findings
As mentioned earlier, oral manifestations of aGVHD are predominated by erythema and ulcerations but can also present as lichen planus–like hyperkeratotic lesions, likely in the context of overlap syndrome. Lip involvement with crusting is also a common feature, similar to that seen in patients with erythema multiforme ( Fig. 1 ).
The diverse spectrum of oral cGVHD manifestations can be classified into 3 distinct groups, according to the main sites and anatomic structures involved: oral mucosal disease, salivary gland disease, and sclerotic disease.
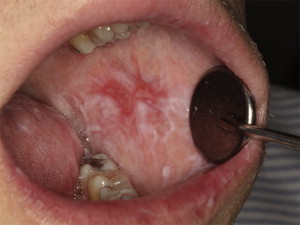
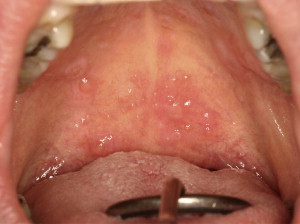
Oral mucosal cGVHD is characterized by lichenoid inflammation that frequently involves the tongue and buccal mucosa but can affect any site in the oral cavity and can range from limited disease with only mild changes to more extensive and symptomatic disease. Clinical changes include white papules, plaques, and hyperkeratotic reticulations resembling Wickham striae in oral lichen planus, erythema, and pseudomembranous ulcerations ( Fig. 2 ). The symptom most often associated with oral mucosal cGVHD is sensitivity to otherwise normally tolerated items, such as spicy, acidic, or rough (crunchy) foods and drinks as well as mint and strongly flavored toothpaste and mouthwashes. Most patients report little or no oral discomfort at rest, even in the presence of extensive oral ulcerations.
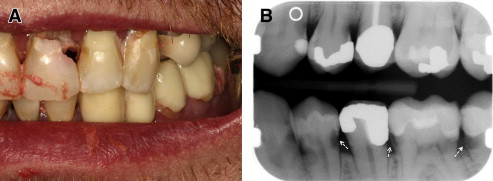
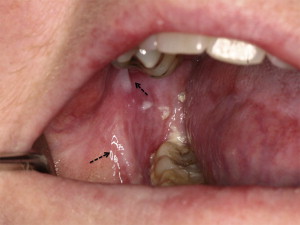
Chronic GVHD of the salivary glands results in both quantitative and qualitative alterations in saliva, including altered concentrations of electrolytes, epidermal growth factor, and salivary proteins. Because of these changes, patients experience increased morbidity from xerostomia, difficulty with speaking, chewing and swallowing, as well as recurrent candidiasis. Patients may present with rampant caries, characterized by cervical and interproximal patterns of decay that develop at a median of less than 2 years after HCT ( Fig. 3 ). In addition to the sialochemical and sialometric changes, oral cGVHD is also associated with recurrent superficial mucoceles, presenting primarily as asymptomatic 0.2- to 0.5-cm vesicles on the palatal mucosa ( Fig. 4 ). Their putative etiopathogenesis is damage to excretory salivary ducts secondary to cGVHD.
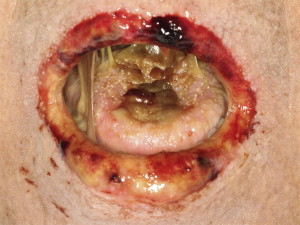
Sclerotic disease affecting the oral cavity is rare but can be a potentially serious complication. Sclerosis can present in the orofacial region as an extension of primary sclerotic cutaneous cGVHD or as a sequela of long-standing mucosal cGVHD and is characterized by sclerodermalike manifestations, including fibrosis and limited mouth opening associated with pain and secondary ulcerations ( Fig. 5 ). The decreased mouth opening results in functional impairment (difficulty with speaking, eating, and maintaining oral hygiene), potentially contributing to infection and malnutrition.
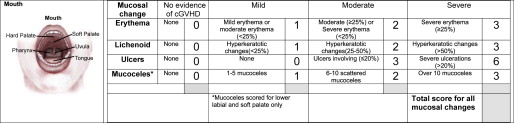
Diagnosis and Assessment
The diagnosis of oral GVHD can typically be made based on history, clinical findings, and context of onset. For standardization purposes, the NIH has introduced criteria for clinical (see Table 1 ) and histologic diagnosis of cGVHD, a scoring system to document the extent and severity of clinical involvement ( Fig. 6 ), and a staging system for assessing the functional impact ( Table 3 ). The global scoring of cGVHD can be calculated based on the number of organs involved (eg, oral cavity) and the severity of involvement according to a 4-point scale, resulting in a classification of mild, moderate, or severe.