Head and neck cancer is a major health concern, with more than 540,000 new cases diagnosed and 271,000 disease-related deaths occurring annually worldwide. As oral and maxillofacial surgeons providing comprehensive surgical management for head and neck cancer, we are continually challenged with difficulty predicting the capricious clinical behavior of head and neck cancer, recurrence at the primary site following resection, cervical and distant metastasis, and the development of second primary head and neck cancers. Early- to moderate-stage oral squamous cell carcinoma (OSCC) is usually treated surgically along with radiotherapy given with or without chemotherapy postoperatively for high-risk patients. In patients with advanced disease, multidisciplinary non-surgical approaches have increasingly been used to improve disease control, survival, and quality of life. However, despite the best combination of surgical and non-surgical approaches, more than 50% of patients with OSCC experience local recurrence or distant metastasis and subsequently have poorer prognoses, and therefore more effective therapies are needed for these patients. Our understanding of the molecular biology of head and neck cancer has progressed significantly over the last decade, and the causes and solutions of these clinical challenges have a molecular basis. The development of novel diagnostic and therapeutic approaches to OSCC requires a better understanding of the molecular pathogenesis of OSCC and the molecular events involved in its growth, invasion, and metastasis. Molecular approaches will greatly increase our ability to predict clinical behavior, determine prognosis, guide surgical treatment, and aid cancer surveillance. This chapter focuses on improving our understanding of the molecular mechanisms of head and neck cancer and its therapeutic implications.
Molecular Biology of Head and Neck Cancer
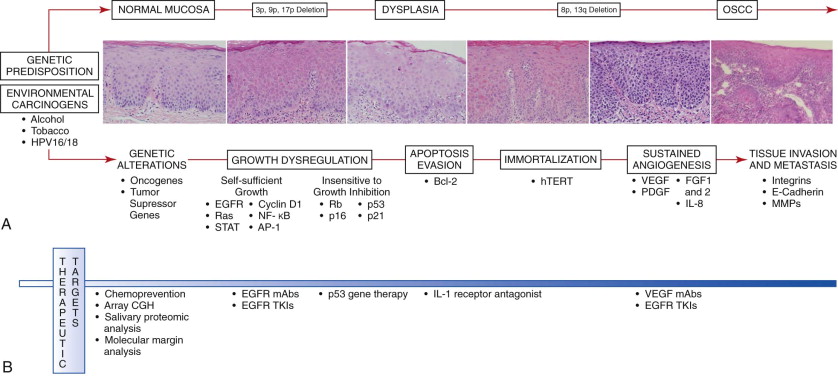
Head and neck cancer is a heterogeneous disease that develops through a complex, multi-step process involving genetic alterations, growth regulation, apoptosis, immortalization, angiogenesis, invasion, and metastasis following a series of molecular events influenced by the individual’s genetic predisposition and environmental exposure to carcinogens ( Fig. 10-1, A ). Chronic exposure to carcinogens such as alcohol, tobacco, betel quid chewing, and oncogenic viruses may result in genetic alterations that can develop into dysplastic lesions and subsequent invasive OSCC. These genetic alterations in oral cancer cells can be divided into two categories. Dominant changes , most frequently occurring in proto-oncogenes, result in gain of function. Recessive changes , or mutations frequently found in genes in the growth-inhibitory pathway or in tumor suppressor genes, lead to loss of function. As a result of these alterations, cancer cells acquire a cellular growth advantage with autonomous growth, evade growth-inhibitory signals, escape apoptosis, and may replicate infinitely. Cancer cells can also stimulate angiogenesis and thereby allow OSCC to further grow, invade, and metastasize. The timing and accumulation of these genetic alterations resulting in carcinoma are probably critical. Because genetic changes precede phenotypic changes, molecular and genetic analysis might afford the clinician the possibility to intervene earlier and improve the prognosis.
Genetic Alterations
Genetic alterations involving the loss of chromosomal material at 3p, 9p, and 17p occurs in a high proportion of dysplastic oral lesions, thus suggesting that these alterations may serve as early markers in oral carcinogenesis, whereas carcinomas more frequently display losses at 8p and 13q and may be associated with later stages of carcinogenesis. The chromosome 3p region includes the tumor suppressor genes FHIT and RSSFIA , which are inactivated by exonic deletion and hypermethylation. Loss of chromosomal region 9p21 occurs in 70% to 80% of dysplastic lesions of the oral mucosa. The CDKN2A locus region of 9p21 encodes the tumor suppressors p16 and p14 ARF , which are often inactivated by promoter hypermethylation. During progression from dysplasia to invasive OSCC, genetic alterations involving the loss of heterozygosity of chromosome region 17p may also occur, as well as p53 gene mutation. Alterations involving p53 in oral dysplasia are associated with increased genomic instability and generally occur late in the progression from dysplasia to invasive carcinoma.
Oncogenes
Proto-oncogenes are highly regulated genes that encode proteins mediating positive cell growth regulation and cell survival signals. If a proto-oncogene is altered via chromosomal translocation, mutation, gene amplification, or retroviral insertion, it may become an activated “gain-of-function” oncogene that promotes uncontrolled cell proliferation and resultant carcinogenesis. Oncogenes may be categorized as (1) growth factors or growth factor receptors, (2) intracellular signal transducers, (3) transcription factors, (4) cell cycle regulators, or (5) those involved in the inhibition of apoptosis. The majority of these oncogenes promote aberrant cell proliferation by overriding various checkpoints of the cell cycle.
Tumor Suppressor Genes
Tumor suppressor genes, or anti-oncogenes, encode proteins that transduce negative cell growth regulation signals such as those involved in cell cycle arrest and apoptosis. In contrast to oncogenes, which are activated by mutation of only one of the two gene copies, tumor suppressor genes are inactivated by point mutations or deletion in both alleles of the gene in a “two-hit” fashion. Once tumor suppressor genes are inactivated, the cell escapes stringent cell cycle control and is predisposed to uncontrolled growth and division. “Loss of function” of multiple tumor suppressor genes is thought to be the major event leading to the development of malignancy.
Field Cancerization
The entire epithelial layer of the oral cavity may be exposed to various carcinogenic insults and is therefore at increased risk for malignant transformation from the accumulation of genetic alterations of oncogenes and tumor suppressor genes. The theory of “field cancerization” has been developed from the finding of dysplastic epithelium adjacent to invasive oral cancers, which accounts for the high incidence of second primary tumors in patients treated for OSCC. In this model, multifocal oral cancers develop from separate, independent genetic alterations, and many of these second primary tumors have been associated with lower survival rates than occurs with the original tumor. In an updated progression model, second or multiple cancers distant from the dysplastic fields have been suggested to be clonally related and derived from the expansion of a common pre-neoplastic progenitor. This occurs when a stem cell located in the basal epithelial layer acquires a genetic alteration and subsequently gives rise to a clonal unit whereby the stem cell and all its daughter cells share the DNA alteration and progress to an expanding field as a result of additional genetic alterations. The resultant mucosal field pushes the normal epithelium aside and may expand to several centimeters in size. These fields may appear as leukoplakia or erythroplakia but often remain clinically undetectable. Clonal selection ultimately results in carcinoma formation within this field of pre-neoplastic cells.
Self-Sufficiency in Growth Signaling
Exogenous growth signals stimulate normal oral keratinocyte proliferation. Growth signals are usually transduced from cell surface receptors that subsequently activate multiple intracellular signaling pathways to result in cell proliferation. This growth signaling may be dysregulated by increases in the level of growth factor receptors or their ligands to promote autocrine stimulation in the absence of exogenous factors during oral carcinogenesis.
Epidermal Growth Factor Receptor
Epidermal growth factor receptor (EGFR) is a member of the membrane-bound receptor tyrosine kinase family. EGF and transforming growth factor-α (TGF-α) are endogenous ligands for EGFR. Following ligand binding, EGFR dimerizes with another EGFR and autophosphorylates, which results in a series of intracellular signaling events that involve activation of the Ras/Raf/mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT), and protein kinase C (PKC) pathways. These signaling events then mediate cell proliferation and survival, invasion, metastasis, and angiogenesis. EGFR overexpression progressively increases from oral premalignant lesions to invasive OSCC. EGFR is overexpressed in 80% to 100% of oral cancers and is an independent prognostic marker that correlates with increased tumor size, decreased radiation sensitivity, and increased risk for recurrence. However, patients with head and neck cancers overexpressing EGFR may exhibit a higher proportion of complete responses to chemotherapy than those with cancers exhibiting low EGFR expression. This overexpression of EGFR with consequently greater intrinsic proliferative activity is thought to result in greater chemosensitivity in cells undergoing mitogenesis.
Ras Oncogene
Ras (H- ras , K- ras , N- ras ) is an important proto-oncogene involved in the regulation of cell growth and transduction of mitogenic cell signaling from the cell surface to the nucleus. The ras gene encodes protein p21, which is constitutively activated through mutation. A high incidence of H- ras mutations has been found in oral cancer, mostly in Asians and possibly associated with betel nut chewing.
STAT Proteins
STAT proteins are latent cytoplasmic transcription factors activated by various extracellular signaling proteins. On activation, STAT proteins can up-regulate the transcription of various target genes and result in uncontrolled cellular proliferation, anti-apoptotic responses, and angiogenesis. Activated STAT3 levels may be elevated in oral cancer via up-regulation of the EGFR, TGF-α, Jak, Src, or interleukin-6 (IL-6) signaling pathways. Activated STAT3 is highly expressed in poorly differentiated oral cancers and is correlated with metastasis and a poor prognosis.
Cyclin D1
Cyclin D1 is a proto-oncogene that regulates the initiation of DNA synthesis. Overexpression of cyclin D1 occurs in a high percentage of premalignant lesions and oral cancers. Cyclin D1 overexpression is an early event in oral carcinogenesis and is associated with more aggressive tumor behavior, an increased rate of lymph node metastases, and a worse prognosis.
Nuclear Factor κB
The nuclear factor κB (NF-κB) is a ubiquitous nuclear transcription factor that regulates many target genes, including immunoregulatory and inflammatory genes, anti-apoptotic genes, and genes that positively regulate cell proliferation. NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), c-Rel, and RelB are included in the NF-κB family. NF-κB expression levels increase gradually from premalignant lesions to invasive cancer. Moreover, NF-κB may inhibit apoptosis through the induction of anti-apoptotic proteins and suppress the apoptotic potential of chemotherapeutic agents, thereby leading to chemoresistance.
Activating Protein-1
The activating protein-1 (AP-1) transcription factor family is made up of multiple Jun (cJun, JunB, and JunD) and Fos (cFos, FosB, Fra-1, and Fra-2) members. AP-1 regulates cellular proliferation, differentiation, apoptosis, oncogene-induced transformation, and cancer cell invasion. AP-1 activation induces transformation and malignant progression in oral cancer; it is found to be activated in both oral dysplasia and OSCC cell lines and is associated with malignant transformation in squamous epithelial cells.
Insensitivity to Growth-Inhibitory Signals
Growth-inhibitory signals are regulated by interactions of cyclin-dependent kinase (CDK), cyclin, and the retinoblastoma ( Rb ) gene product. The proteins encoded by the tumor suppressor genes p16 , p15 , p21 , and p53 also act as inhibitors of cell cycle progression, and when their expression is lost, progression through the cell cycle is increased. Loss of growth-inhibitory signals and the development of self-sufficient growth signals via oncogene activation are needed for oral carcinogenesis.
Retinoblastoma Gene
The Rb gene product is a crucial regulator of G 1
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


