Introduction
The aims of this study were to describe bacterial load and diversity of the aerosol created during enamel cleanup after the removal of fixed orthodontic appliances and to assess the effect of a preprocedural mouth rinse.
Methods
The study involved the sampling of ambient air adjacent to the patient’s mouth during adhesive removal using a slow-speed handpiece and a spiral fluted tungsten carbide bur without water irrigation. Sampling was carried out during enamel cleanup with or without a preprocedural mouth rinse of either sterile water or chlorhexidine. Airborne particles were collected using a viable inertial impactor simulating the human respiratory tree. The bacteria collected were analyzed using both culture and molecular techniques.
Results
Bacteria produced during debond and enamel cleanup can reach all levels of the respiratory tree. The use of a preprocedural mouth rinse, either sterile water or chlorhexidine, increased the numbers and diversity of the bacteria in the air.
Conclusions
When using a slow-speed handpiece and a spiral fluted tungsten carbide bur for enamel cleanup after orthodontic treatment, the bacterial load and diversity of the aerosol produced are lower when a preprocedural mouth rinse is not used.
Highlights
- •
Slow-speed enamel cleanup creates a bioaerosol that infiltrates deep into the lungs.
- •
A water or chlorhexidine rinse increases the numbers and diversity of airborne bacteria.
- •
Bacteria were still found in the air at each impactor stage, even without enamel cleanup.
In the dental clinic, many instruments can generate aerosol particulates: ie, those with aerodynamic diameters of 50 μm or less. These include air turbines, ultrasonic scalers, and slow-speed handpieces, all of which may be used during enamel cleanup after orthodontic debond. Whereas large particles created during their use may show a ballistic trajectory and fall quickly to the floor or other nearby surface, smaller particles can remain airborne for extended periods and therefore be inhaled by dental operators, assistants, and patients.
Once inhaled, the level at which these particles settle in the respiratory tree depends on factors such as lung anatomy, nasal or oral breathing pattern, the presence of respiratory disease, particle size, and particle density. Although many settle superficially in the conducting airways and terminal bronchi, smaller particles may impact as deep as the terminal alveoli and be retained in the lungs for long periods before being cleared by the alveolar macrophages.
In addition to solid particulates created during operative dental procedures, liquid aerosols may also be created. Such aerosol droplets may contain solid particulates as well as microbial contaminants—bacteria and fungi—in which case they are often referred to as bioaerosols. The dynamics of aerosol behavior are complex and depend on factors such as liquid composition and environment, including temperature, humidity, and air flow. A previous study showed that bioaerosols have considerable potential for infection of human hosts when the droplet diameter is 0.5 to 20 μm.
Interestingly in dentistry, orthodontists perceive themselves to be at less risk of contracting diseases from their patients than do general dental practitioners, probably because their patients tend to be predominantly children and young adults. However, perhaps as a consequence of this perceived lower risk, orthodontists are less likely to adhere to infection control procedures. This is despite the fact that within 5 minutes of beginning enamel cleanup at orthodontic debond, there is a significant increase in bioaerosols compared with preoperative resting room levels. The aerosol particles liberated into the working atmosphere contain microorganisms from the patient’s oral cavity.
Although microbiological culture techniques have previously been used in the examination of dental aerosols, approximately 50% of intraoral microbes cannot be cultured in the laboratory. Polymerase chain reaction (PCR) and denaturing gradient gel electrophoresis (DGGE) techniques can be used to identify additional microbial species, but to date no published studies have used these techniques in the microbiological analysis of dental or orthodontic aerosols.
The aim of this study was to investigate the bioaerosols created during the debonding and enamel cleanup after orthodontic fixed appliance therapy. The specific objective was to investigate the effect of preprocedural rinsing before debond, with either water or chlorhexidine gluconate, on the bacterial load and biodiversity of the aerosol produced.
Material and methods
Eighteen consecutive patients undergoing routine debonding of their maxillary and mandibular fixed orthodontic appliances followed by enamel cleanup were recruited into this study. Eligible patients were those who had received comprehensive fixed appliance treatment: the maxillary and mandibular arches were treated to completion, and the patient was scheduled for debonding of the fixed appliances. Patients undergoing single-arch or sectional appliance treatment were excluded, as were those who were having or recently had antibiotic treatment. Selection for inclusion in the study was on a consecutive basis if the eligibility criteria were met. No age restrictions were set, nor were there maximum or minimum limits on treatment time. The patients were divided into 3 groups of 6, with each group allocated to a particular method of composite removal at appliance debond: (1) slow-speed handpiece, no preprocedural mouth rinse; (2) slow-speed handpiece, 0.2% chlorhexidine gluconate preprocedural mouth rinse; and (3) slow-speed handpiece, sterile water preprocedural mouth rinse. All 6 patients in a group were sampled before proceeding to the next group.
Formal ethical committee approval was not required since this was not an interventional study, but a study of air quality. All 3 composite cleanup methods were already in routine use at debond in the orthodontic department at Musgrove Park Hospital (Taunton, Somerset, United Kingdom). Before debond, each patient’s oral hygiene status was recorded using the Index of Oral Cleanliness, a 5-point scale proposed by Bearn et al.
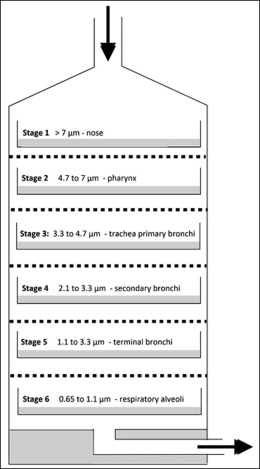
Air sampling was carried out in the orthodontic department using a Thermo-Electron 10-800 6-stage viable particle sampler (Thermo Fisher Scientific, Richardson, Tex) ( Fig 1 ). This type of impactor has been in use since the early 1980s and comprises 6 impactor stages that correspond to various levels in the respiratory tree, as shown in Figure 2 .

Each of the 6 stages of the impactor contained a glass petri dish in which there was a solid fastidious anaerobe agar containing 5% defibrinated horse blood. Attached to the top of the impactor at the inlet port was an extension tube used to simulate the pharynx. Attached to the bottom of the impactor was a vacuum pump used to draw the air through the impactor stages. The air flow through the impactor was set at 28.3 liters per minute according to the manufacturer’s instructions. The air intake of the extension tube was positioned so that it was level with the patient’s mouth at debond and at a distance of 30 cm.
In patient groups 2 and 3, each patient was initially asked to perform a preprocedural mouth rinse. The preprocedural chlorhexidine and water mouthwashes were dispensed in plastic cups in volumes of 15 mL, and the patients were asked to rinse thoroughly for 1 minute, just before bracket removal.
In each group, the maxillary and mandibular fixed appliances were removed using debanding and debonding pliers as is normal practice. After this, the impactor was allowed to run for 1 minute before composite removal. Air sampling was then carried out for 15 minutes during the composite removal procedure. If composite removal was completed in less than this time, the impactor was still allowed to run for the full 15 minutes. For each patient, composite removal was performed using a spiral fluted tungsten carbide bur in a slow-speed handpiece without water coolant, but with a high-volume chairside aspirator held close to the patient’s mouth as is normal practice. For all patients, the composite that had been used to bond the brackets to the teeth and was being removed at debond was Transbond XT (3M Unitek, Monrovia, Calif). The band cement was Intact Glass Ionomer Cement (Orthocare, Bradford, United Kingdom).
After debond, composite removal, and sampling, the 6 glass petri dishes were removed from the impactor, covered, and transferred to an anaerobic incubator for 7 days at 37°C. After this incubation period, the dishes were removed and under good illumination examined for microbial growth on the agar surface. For each petri dish, the colony counts and numbers of different colony types were noted, along with the total numbers of colony-forming units for each type. In distinguishing 1 colony type from another, factors such as size, color, surface texture, dryness or wetness, and contour of the colony margins were examined with the naked eye.
Finally, the total bacterial growth on each plate was collected by washing with 1.5 mL of Tris-HCl/EDTA buffer solution and sweeping the surface with sterile plastic loops to collect all adherent colonies. This plate sweep sample was pipetted into 2.0-mL Eppendorf containers and stored at −80°C in a laboratory freezer for later molecular analysis.
The molecular techniques applied to each plate sweep sample were (1) extraction of DNA; (2) “universal” 16S rDNA-directed PCR of the extracted DNA, using a thermocycler; (3) agarose gel electrophoresis of the PCR product; and (4) DGGE of the PCR product.
Once each petri dish had been swept for bacterial growth, the growth medium was removed, and the dish thoroughly cleaned and sterilized before reuse.
For each patient sample, 6 Eppendorf tubes were removed from the freezer and left to defrost on the laboratory workbench at room temperature for 20 minutes. After defrosting, the GenElute kit (Sigma-Aldrich, St Louis, Mo) was used to extract the bacterial DNA. The sample was subjected to a number of standardized chemical processes designed to extract the DNA from gram-positive bacterial cells: (1) bacterial breakdown to release DNA, (2) preparation of a DNA-binding membrane, (3) transfer of the bacterial lysate through the membrane to retain the DNA, and (4) washing of the membrane to release the DNA.
These stages were performed according to the manufacturer’s instructions, and all chemicals required were included in the standard kit as supplied. Other materials necessary were routine items of equipment found in most microbiology laboratories, including a centrifuge, hot water baths, and a vortex mixer. The result of this process was a solution containing the collected bacterial DNA of all species for each petri dish sweep sample.
A PCR stage was required to amplify the extracted DNA to a level detectable by DGGE. For a set of 6 DNA samples corresponding to the collected bacteria at each of the 6 impactor stages, a stock solution was prepared consisting of (1) PCR water (distilled), 88.5 μL; (2) green buffer, 60 μL; (3) forward primer, 30 mmol/L 12 μL (27F: AGA GTT TGA TCC TGG CTC AG); (4) reverse primer, 30 mmol/L 12 μL (1492R: TAC GGG TAC CTT GTT ACG ACT T); (5) dNTP mix, 6 μL; and (6) Taq polymerase, 1.5 μL.
This was dispensed into 6 small Eppendorf tubes in volumes of 30 μL, labeled to indicate impactor stages 1 to 6, and an extra seventh tube was prepared with these components in the same proportions. To the seventh tube, 20 μL of distilled PCR water was added, and all 7 tubes were subjected to 1 minute of ultraviolet light exposure in a hooded ultraviolet light transilluminator. This stage was carried out to eliminate any viable DNA that may have contaminated the Eppendorf tubes during the preparation process. To the first 6 tubes, 20 μL of DNA was added from each of the 6 impactor stage samples, and the seventh tube was retained as a negative control. The 7 Eppendorf tubes were placed in a PCR thermocycler and subjected to the following: (1) predenaturation step at 94°C for 2 minutes; (2) 34 cycles of 94°C for 30 seconds, 56°C for 60 seconds, and 72°C for 2 minutes; (3) final extension step of 72°C for 10 minutes; and (4) temperature held at 10°C. To confirm successful DNA extraction, the amplified PCR product was run on an agarose gel.
A 1.2% concentration of agarose gel (Gene Flow Ltd, Lichfield, Staffordshire, UK) was prepared by adding 1.2 g of agarose powder to 100 mL of Tris-Borate-EDTA (Fisher Scientific, Louhgborough, UK) in a 100-mL beaker and heating the mixture in a microwave oven for 2 minutes at 800 W. The beaker was then placed on a magnetic stirrer and allowed to cool until it began to thicken. At this point, 3 μL of ethidium bromide was added and allowed to disperse under the action of the magnetic stirrer. An agarose gel casting kit tray was prepared according to the manufacturer’s instructions, with a comb insert creating sample wells. This was removed after cooling.
An agarose marker solution was created by adding 15 μL of a 100 base-pair ladder to 50 μL of loading dye, and 6 μL of this was pipetted into the first well; this also acted as a positive control. Into adjacent empty wells, 10 μL of the PCR product for each impactor stage was pipetted, and the position of each sample in the gel was carefully recorded. The gel tray was then seated in an electrophoresis cell and the tank filled with TBE buffer. Once immersed, the tank was closed, and 120 V of electrical potential was applied for 45 minutes across the gel.
Removing the gel from the tray and exposing it to ultraviolet light in the transilluminator allowed confirmation of a positive PCR product by noting the dark bands far from the sample wells. Image capture was performed using the DigiDoc-It Darkroom bioimaging system (UVP, Upland, Calif).
After confirmation of the PCR product, the DGGE step was required to identify the different bacterial species in a sample obtained from each stage in the impactor. This required the use of a gel chamber into which 2 solutions would be combined as liquid but would set to form a 0.75-mm-thick rectangular gel, in which the concentration of urea formed an increasing gradient from top to bottom.
The solutions were prepared as follows.
Solution A was (1) 11.5 mL of 70% Denaturing Gradient Gel Electrophoresis (DGGE) denaturant (stock solution of 18.8 mL of 40% acrylamide, 2 mL of 50 times Tris-acetate Ethylenediaminetetraacetic acid (EDTA) buffer, 16 mL of formamide, 16.8 μL of urea, made up to 100 mL with distilled water); (2) 80 μL of Ammonium Persulfate; and (3) 5 μL of Tetramethylethylenediamine.
Solution B was (1) 11.5 mL of 40% DGGE denaturant (stock solution of 18.8 mL of 40% acrylamide, 2 mL of 50 times Tris-acetate EDTA buffer, 28 mL of formamide, 29.4 μL urea, made up to 100 mL with distilled water); (2) 80 μL of Ammonium Persulfate; and (3) 5 μL of Tetramethylethylenediamine.
The gel was prepared by the addition of solutions A and B to the 2 separate chambers of a gradient maker, which controlled the ratio of 1 solution to the other and allowed a steadily changing mixture of the 2 solutions to flow into the glass plate assembly via a blood collection kit. Throughout this process, a magnetic stirrer ensured that the A and B mixture was homogenous. The combined denaturants were run into the gel chamber until the top had been reached. A plastic comb was inserted into the liquid to form a number of wells once the gel had set. The gel was allowed to set for 20 minutes.
Once set, the comb was removed to expose a number of open wells on the upper edge of the gel. For each sample, 6 μL of the PCR product was combined with 6 μL of the DGGE loading dye; 10 μL of this mixture was pipetted into an empty well at the top of the gel, and the position of each sample was recorded. The remaining 2 μL of the mixture allowed for a margin of pipetting error.
The gel chamber was lowered into the DGGE electrophoresis tank (Model no 2401, CBS Scientific Co, Del Mar, California, USA), containing 22 L of Tris-acetate EDTA, maintained at 60°C, and 220 V was applied through the gel for 4 hours. Once complete, the chamber was removed and disassembled.
A solution of Sybr Green staining reagent (ThermoFisher Scientific, Basingstoke, UK) in 1 times Tris-acetate EDTA was prepared in the ratio of 15 μL Sybr Green to 150 mL of Tris-acetate EDTA. The DGGE gel was transferred from the gel chamber to a plastic bath containing this staining solution, which was then placed on an oscillating rocker table for 20 minutes. After 20 minutes of gentle agitation, the staining stage was complete, and the gel was transferred to the transilluminator and exposed to transilluminated ultraviolet light. Once a sequence of bands in the gel had been visualized, the digital science electrophoresis documentation and analysis system (Kodak, Rochester, NY) was used to record the resultant image.
On each image, the numbers of unique bands (ie, different bacterial species) per impactor stage were noted, providing data on the biodiversity of the samples obtained under the 3 sampling conditions.
Statistical analysis
Data analysis was undertaken on a descriptive basis. To describe the bacterial load and biodiversity means, standard deviations are provided.
Results
Six patients were recruited for each of the 3 debonding groups (no rinse, chlorhexidine rinse, and water rinse), with each patient having 6 samples, one for each impactor cutoff stage. With respect to the Index of Oral Cleanliness, as might be expected of patients undergoing orthodontic treatment, few patients scored highly: ie, they had poor oral hygiene. Most patients scored 0, 1, or 2, with only 1 patient having a score of 4, and this patient had no rinse before debond.
Table I summarizes the bacterial load for each of the 6 impactor stages at background level and in each patient group. In all 3 groups, the total bacterial count was higher than at background level. For each group and at background, the lowest count was recorded at impactor stage 6 (corresponding to the level of the respiratory alveoli), and the highest levels were from levels 2 to 5 (corresponding to the pharynx down to the terminal bronchi, respectively). Preprocedural rinsing with either chlorhexidine gluconate or water was associated with an increased bacterial load.


