The spectrum of facial clefting malformations encompasses a group of conditions, including mandibulofacial dysostosis. Within this group are Treacher Collins syndrome, Nager syndrome, Miller syndrome, and others. These rare conditions are predominantly autosomal dominant and have varying degrees of expressivity and severity. The conditions are always bilateral, and their expression is typically asymmetric. The cause is unknown, but the pathogenesis has been described in animal models with phenotypes similar to the condition. In animal models, the conditions can be produced by vascular insults to the stapedial artery and by injection of retinoids at specific embryonic intervals.
The lateral parts of the face are the targets of these conditions, with the orbits, eyelids, lateral canthus, zygoma, mandibular ramus, skin of the cheek, muscles of mastication, and external and middle ear structures being affected. Displaced hair from the eyelashes and scalp is often observed on the cheek. Because of the hypoplasia of the mandible, the upper airway is commonly narrow at the base of the tongue. Compounding the airway obstruction, choanal atresia can also be observed.
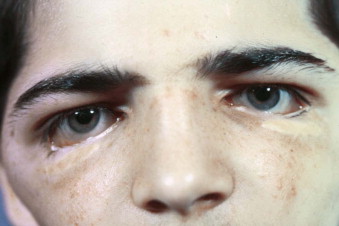
The facial phenotype is classic and consists of down-turned lateral palpebral fissures, lateral upper eyelid ptosis, coloboma of the eyelids, lateral and inferior orbital wall hypoplasia, proptosis, overprojection of the nasal dorsum, narrow middle face, short posterior face, elongation of the anterior face, mandibular hypoplasia with retrogenia, and lip incompetence.
The oral findings are variable and include an open bite, cleft palate, and flared mandibular anterior teeth. The occlusion varies depending on the expression of the condition. Apertognathia is also variable. Although mandibular retrognathism and retrogenia are typical, paradoxic class III malocclusion can be observed.
Radiographic findings include a short anterior cranial base, egg-shaped orbits oriented obliquely from medial to lateral, clefts of the zygomatic arch, hypoplastic or aplastic zygomas and mandibular condyles, short ramus height, antegonial notching, retrognathism, and retrogenia. The occlusal and mandibular planes are always steep.
Functional Findings
Hearing
Conductive hearing loss almost always accompanies the condition. The external and middle ear structures are usually involved. For this reason, newborn hearing screening should always be performed, and when abnormal findings are encountered, conductive hearing aids should be applied as soon as possible. Early intervention may improve learning, expressive language, and other communication skills. The middle ear structures can be missing or ankylosed, the ear canal can be atretic or hypoplastic, and the tympanic membrane can be absent. The external ear may also be malformed, ranging from almost complete absence to hypoplasia of the entire external structure.
Respiration
Newborns affected with mandibulofacial dysostosis commonly have respiratory difficulty. This stems from hypoplasia of the nasopharynx and obstruction of the base of the tongue caused by mandibular hypoplasia. The majority respond favorably to prone posturing and avoidance of the supine position. Tracheostomy is an option, but the decision to proceed with it should not be taken lightly. Perinatal tracheostomy may be lifesaving, but postsurgical mortality (up to 15%) can occur with this procedure in neonates and young children. In addition, it places an enormous burden and pressure on families, hospitals, and nursing staff to clean and maintain these devices. When the airway problem is isolated to the base of the tongue and not associated with laryngomalacia, tracheomalacia, or other airway problems, distraction osteogenesis of the mandible is an alternative. This decision, too, should not be taken lightly since it is not a panacea and it will have adverse consequences on the developing dentition, will cause scars, and will require care provider support to activate and keep the appliances clean. Other soft tissue procedures to improve the airway have been described, such as lip-tongue adhesion, but the results are inconsistent. If choanal atresia or hypoplasia accompanies the condition, the nasal airway is additionally compromised. Surgery to relieve the area of obstruction is the only means of treating this condition. The best timing for this surgery is debatable.
Feeding
Swallowing problems are not typical of this condition; however, most affected neonates have eating problems because of the limited nasal and oropharyngeal space. Neonates are obligate nasal breathers and rely on sucking to assist in the ingestion of milk and fluids. With the airway compromise associated with mandibulofacial dysostosis, feeding is always an issue, and it requires more time and energy than normal. Tube feeding to supplement oral feeding is occasionally helpful. Gastrostomy is an alternative but should be reserved for those with swallowing disorders or failure to thrive. Gastroesophageal reflux is another potential contributor to feeding and airway issues in these patients, and occasionally fundoplication is necessary. Nursing staff and parents must be trained to be patient with feeding.
Speaking
Cleft palate occasionally accompanies the mandibulofacial dysostosis conditions. When it does, a cleft palate protocol should be used, except when airway concerns supersede the routine protocols. In general, as the infant gains weight and grows, the mandible advances and the oral pharyngeal space opens, thereby permitting the palate to be repaired at 9 to 18 months of age.
Children with Nager syndrome characteristically have a flaccid, paralyzed, or aplastic soft palate. This condition is one of the most frustrating aspects of cleft care since even obliteration (either surgical or obturation with a prosthesis) has little effect on the hypernasal speech.
When children with mandibulofacial dysostosis have speech problems and the palate is not cleft, it is more likely to be due to hearing issues rather than velopharyngeal dysfunction.
Vision
Many patients with mandibulofacial dysostosis wear corrective lenses. Strabismus, amblyopia, and other visual impairments can accompany these conditions. Other than occasional coloboma of the iris or pupil (or both), there is no typical pattern of visual disturbances with these conditions. Attaching hearing aids to the eyeglass frame avoids wearing a headband to support the aids and encourages the use of both devices simultaneously.
When orbital surgery is performed, diplopia is expected postsurgically, but it usually subsides as the swelling resolves. The diplopia subsides even when bone grafts are placed in the lateral orbital floor to change the shape of the orbit and elevate the level of the globe.
Jaw Function
Ankylosis of the temporomandibular joint has been reported with this condition, but it has never been observed by this author. Most commonly, patients are able to open and close the jaw. Because the muscles of mastication are affected to varying degrees, an inability to excurse laterally or to protrude symmetrically is commonly observed. Reduced biting force is expected because of the abnormal masseter, pterygoid, and temporalis muscles.
Even in the absence of normal temporomandibular joints and with compensatory recruitment of parafunctional muscle activity to open and close the jaw, myofascial pain is infrequent in these conditions.
An inability to incise or masticate efficiently is more associated with malocclusion than with abnormal jaw function.
Surgical Intervention
ZygomaticOorbital Reconstruction
Surgery can improve the appearance and function of patients with these conditions. The protocol used depends on the expression of the condition. Surgery during infancy should be undertaken to preserve life and vital function. Airway and vision are the two issues that require early intervention. Tracheostomy, distraction osteogenesis, gastrostomy tube placement, fundoplication, choanal atresia repair, and eyelid surgery should be considered to preserve life and vision. If the extent of eyelid absence results in drying and repetitive corneal ulcers, early eyelid reconstruction is indicated. In the absence of globe trauma or drying, eyelid surgery should be reserved for post-skeletal reconstruction. The scarring secondary to eyelid surgery produces scars that can be unsightly ( Fig. 95-1 ).
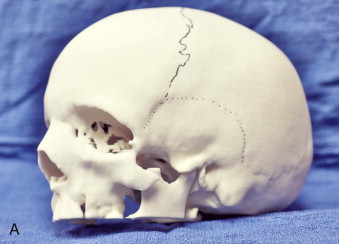
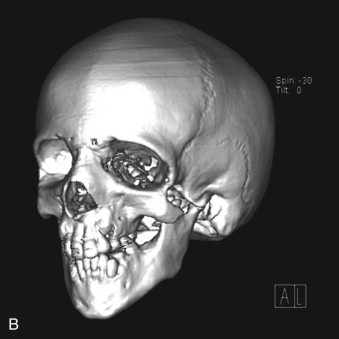
The initial phase of reconstruction commences at approximately 8 years, depending on the patient and parents’ wishes. Orbital and zygomatic reconstruction can be performed with full-thickness temporoparietal bone grafts (large—10 cm). Square blocks can be harvested by conventional neurosurgical methods via burr holes and a craniotome. Selection of the donor site is based on the curvature of the skull and thickness of the bone. Presurgical planning with stereolithographic models, three-dimensional computed tomography (CT), or virtual planning can help determine the best donor site and the precise shape of the defect to be reconstructed ( Fig. 95-2 ).
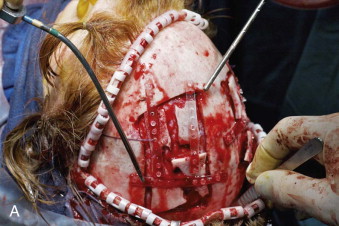
A coronal incision is used to approach the orbits, zygoma, and lateral orbital walls. The calvarial bone grafts are harvested through this same incision. Once these grafts are harvested, the exact size and shape can be carved from the block. The remaining pieces are then split and the donor site is reconstructed with the remnants. In general, biodegradable mesh is used to reassemble the pieces and to attach the construct to the skull ( Fig. 95-3 ). Through the same incision, the dissection continues to the forehead and orbital region. It is important that the dissection remain subperiosteal over the forehead area and in the region of the zygomas to prevent injury to the facial nerve.
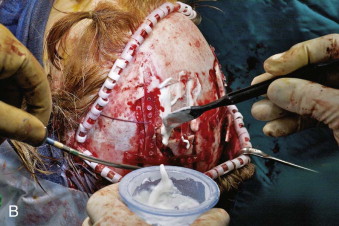
The temporalis muscle is usually intact unless it is elevated during harvest of the cranial bone. The plane of dissection should be under the temporoparietal fascia and either just superficial to or below the superficial layer of the deep temporal fascia. Doing this avoids damage to the facial nerve. This plane of dissection is the same as the subperiosteal plane of the orbit and lateral cheek. The dissection is carried around the infraorbital rim and the lateral maxilla adequately enough to permit insertion of the bone grafts. Subperiosteal dissection of the orbit is limited to the roof, lateral walls, and floor of the orbit. The lateral canthus is always detached. Frequently, the inferior orbital fissure has no anterior or lateral component because of the missing or hypoplastic zygoma. Dissection of tissue over the mid-face must be extensive enough to permit insertion and coverage of the bone graft. Scoring the periosteum with a scalpel helps stretch the soft tissues.
The bone graft is crafted according to the template taken from the stereolithographic model or constructed from virtual planning ( Fig. 95-4 ). The bone graft is inserted and positioned as planned and is then secured with bone screws by anchoring the graft to the lateral orbital wall and to the temporal bone posteriorly. The floor of the orbit on the lateral aspect is usually grafted as well. Once both grafts are secured, the coronal flap is repositioned. The lateral canthus is identified, elevated, and secured to the temporalis fascia with a permanent suture. The flap is then closed in layers while being certain to resuspend the fascial layers to the temporalis fascia or to the calvaria via small burr holes through the outer cortex.
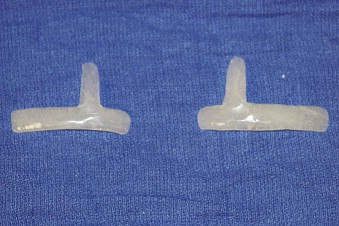
Chin Reconstruction
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


