The group of diseases now collectively known as Langerhans cell histiocytosis (LCH) include entities formerly referred to as histiocytosis X, eosinophilic granuloma, Letterer-Siwe disease, Hand-Schüller-Christian disease, Hashimoto-Pritzker syndrome, self-healing histiocytosis, pure cutaneous histiocytosis, Langerhans cell granulomatosis, type II histiocytosis, and the generic term nonlipid reticuloendotheliosis. LCH is a rare disease that affects five children per million population and about one to two adults per million population. It is predominantly a childhood disease, with more than 50% of affected individuals younger than 15 years of age. The peak incidence is between 1 and 4 years old. The first case of LCH was described by Dr. Thomas Smith in 1865 in a 4-year-old with punched-out lesions of the calvarium. The skull defects were originally thought to be congenital based on drawings because x-rays were not developed until 1895. The child died 1 month later of respiratory complications related to whooping cough. Hand-Schüller-Christian disease was independently described by Hand in 1893, Schuller in 1915, and Christian in 1920 in patients with exophthalmos, polyuria, and skull defects. The disease was initially attributed to tuberculosis infection. Letterer-Siwe disease was described by Letterer in 1924 and by Siwe in 1933 as an acute disorder of the reticuloendothelial system in two young children. This disease was characterized by marked hepatosplenomegaly, lymphadenopathy, bone tumors, anemia, and hyperplasia of nonlipidized histiocytes. Eosinophilic granuloma of bone was first described by Mignon in 1930 as a granulomatous bone lesion in an adolescent male.
In 1940 Lichtenstein and Jaffe described a case of a solitary bone lesion of the femur in a 4-year-old girl that did not suggest any classified disease of bone, either inflammatory or neoplastic. This case also notably consisted of sheetlike collections of histiocytes. Earlier reports suggested the possibility of myeloma with prevalence of eosinophilic cells or osteomyelitis with eosinophil reaction. The authors believe that the lesion represented neither a myeloma nor an osteomyelitis as had been suggested by others, but rather some type of granuloma. However, it did not fit into any of the recognized granuloma categories. Moreover, they proposed that the granuloma existed in response to a virus. The name eosinophilic granuloma of bone was applied because of the preponderance of eosinophils, although the authors fully recognized that the basic cells were histiocytes. The authors pointed out those specific clinical features that might lead one to suspect the presence of eosinophilic granuloma of bone even before surgical intervention is attempted. Among these were the occurrence of the lesion in a child or a very young adult; a short history of painful localized swelling of only several weeks duration; radiographic evidence of a rarefied and destructive bone lesion, especially in the calvarium, but also in the ribs or in a long bone and with perforation of the cortex; and perhaps a palpable soft tissue mass overlying the affected bone. Jaffe and Lichtenstein were quick to point out that these features may lead the clinician to suspect malignant disease, specifically a Ewing’s sarcoma.
Jaffe and Lichtenstein also reported the realization that multiple bone lesions may be encountered and of the same kind as the solitary lesions. They recommended that even in cases in which there was supposedly only a solitary lesion in bone, additional lesions may be present and should be searched for radiographically. This radiographic investigation is referred to as a skeletal survey. Aside from the known focus of LCH, a negative skeletal survey in combination with no suspicion for visceral involvement will proclaim a patient has monostotic eosinophilic granuloma.
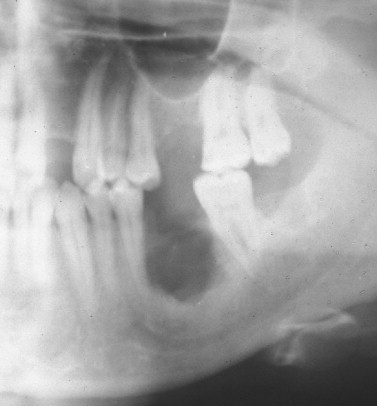
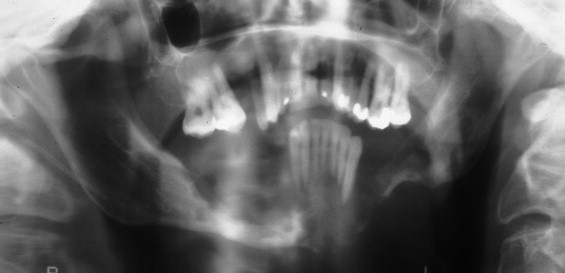
The clinical varieties of LCH range from a curable, solitary, destructive lesion of bone to a lethal leukemia-like disorder that primarily affects infants. The clinically evident pathologic condition in LCH is broadly divided into two categories: direct involvement with the disease (i.e., bone or organ lesions) and secondary consequences resulting from permanent damage by the primary disease (i.e., exophthalmos, diabetes insipidus, fractures, and loss of teeth). This pathologic process is well recognized to be a neoplastic process. LCH affects the head and neck region quite commonly, particularly the skull and jaws. With jaw involvement, 90% of patients are younger than 40 years of age, with a mean of 19 years. Unifocal involvement of the jaws represents about 50% of maxillofacial LCH, with multifocal single system disease and multifocal multisystem disease equally accounting for the other 50%. The mandible is involved approximately three times more commonly than the maxilla. The posterior region of the jaws is more commonly involved than the anterior region. Early disease in the oral and maxillofacial region is manifested by lytic lesions of the jaws, often seen as localized, punched-out radiolucencies with no calcification and no sign of sclerosis or reaction at the borders ( Figure 28-1 ). The most common symptom is pain, although a mass may also be a presenting symptom. With advanced disease, there may be severe alveolar bone resorption producing the appearance of “teeth floating in air” ( Figure 28-2 ). As will be discussed in detail in this chapter, the extent of the disease process often dictates surgical therapy for a unifocal eosinophilic granuloma of the jaws. Prompt diagnosis of disease is paramount to providing curative surgical therapy with little or no deformity. The surgeon should consider LCH on a differential diagnosis of lucent lesions of the jaws, especially in patients younger than 40 years of age.
THE LANGERHANS CELL
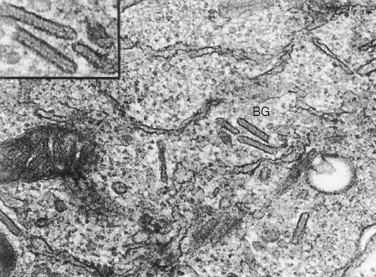
The term histiocyte refers to two groups of immune cells: macrophages and dendritic cells. The Langerhans cell is a mononuclear cell of bone marrow origin that belongs to the dendritic cell family. It can be generated from peripheral blood monocytes by the action of granulocyte-macrophage colony-stimulating factor and tumor necrosis factor ∝. The normal Langerhans cell is recognized to be an important antigen-presenting cell in the skin that represents the most peripheral extension of the immune system. They are normally found in mucosa, lymph nodes, and bone marrow. With the uptake of antigen, Langerhans cells migrate to afferent lymphatic channels and make their way to paracortical zones in regional lymph nodes. Once there, the endocytosed antigens are presented to naive helper T cells. Langerhans cells have not proven to be clonal. In 1868 Paul Langerhans, a 21-year-old medical student at the University of Berlin under the mentorship of Professor Virchow, described the cell that bears his name. His initial impression was that the cell was a nerve receptor cell because of its stellate, dendritic appearance. Only in later years did Langerhans realize that his original supposition was incorrect. In 1961 Birbeck et al described the organelle that has become the unique label of the cell. Interestingly, there are stages of the cell’s development when the Birbeck granule is not identified. Typically, two characteristics are observed in Langerhans cells. The first is the presence of Birbeck granules. These intracellular structures are rod shaped with a ballooning at one end that resembles a tennis racket. Under electron microscopy, the Birbeck granule ( Figure 28-3 ) consists of a bilayered membrane. They can also be observed to develop as invaginations of the cell membrane. These findings resulted in a hypothesis that the granules represented the cellular response to an antigen presented on this cell and therefore reflect the normal function of Langerhans cells. One theory with respect to the pathogenesis of LCH therefore is that immunologic stimulation of this normal antigen-presenting cell may continue in an uncontrolled manner, and this results in proliferation and accumulation of these cells. The second characteristic of Langerhans cells, which is diagnostic of the pathologic condition, is that they stain strongly positive for the presence of CD1a on their surfaces. This property is highly characteristic of Langerhans cells, but not of other cells of histiocytic origin.
CLASSIFICATION OF LCH
In 1953 Lichtenstein lamented that his 1944 paper did not offer acceptable nomenclature for his discovery published in 1940. He believed that some designation built around histiocytosis was appropriate since this term had the reference to an inflammatory proliferative reaction and it was the one feature common to all of the various pathologic expressions of the disease. The designation, idiopathic histiocytosis , was considered, but discarded by Lichtenstein because it lacked specificity in that it might also apply to lesions of sarcoidosis. Therefore he presented “histiocytosis X” for the disease under consideration, and suggested the classification seen in Box 28-1 in 1953. The designation histiocytosis X was proposed because of brevity and the implication of the need to search for the etiologic agent. In 1953 Lichtenstein reorganized his thoughts on histiocytosis X and proposed a more simple classification in his landmark paper, as seen in Box 28-2 . This classification is widely accepted in the twenty-first century, although under the designation, Langerhans cell histiocytosis. A writing group of the Histiocyte Society was created that developed a classification as a standard for diagnosis and patient management in cases of Langerhans cell histiocytosis. Their initial publication reinforced the correct nomenclature for this disease process as Langerhans cell histiocytosis rather than histiocytosis X as had previously been standard. The most current method of categorization of LCH is based on risk stratification of affected individuals into recommended treatment protocols formulated by the Histiocyte Society ( Box 28-3 ). Unifocal disease involves a single disease system with a single site of involvement. There is a good prognosis in this group, and older children and adults are typically affected. Multifocal single system disease most commonly affects young children and is present when several lesions are identified in a single organ system. Bone is most commonly involved, and this represents multifocal eosinophilic granuloma. The prognosis is intermediate. Multifocal multisystem disease carries the worst prognosis where multiple lesions are found in more than one organ group. Commonly affected organ systems include bone, skin, liver, spleen, and lymph nodes. Children younger than 2 years of age represent the majority of patients in this group.
-
Histiocytosis X, localized to bone (eosinophilic granuloma, solitary or multiple)
-
Histiocytosis X, disseminated, acute, or subacute (Letterer-Siwe disease)
-
With destructive skeletal lesions (eosinophilic granuloma)
-
With transition to chronic phase (Hand-Schüller-Christian disease)
-
Histiocytosis X, disseminated, chronic (Hand-Schüller-Christian disease)
-
With destructive skeletal lesions (eosinophilic granuloma)
-
With early extraskeletal lesions (indicate sites) resembling eosinophilic granuloma
-
With acute or subacute exacerbations (Letterer-Siwe disease)
-
With involvement primarily of bone, lungs, pituitary, and/or brain, skin, mucous membranes (oral, anal, genital), liver, or lymph nodes, etc. (in varying combinations)
-
Monostotic or polyostotic eosinophilic granuloma of bone —solitary or multiple bone lesions without visceral involvement.
-
Chronic disseminated histiocytosis —a disease involving bone, skin, and viscera. This is referred to as Hand-Schüller-Christian disease, and may involve a triad consisting of punched-out calvarial lesions, exophthalmos, and diabetes insipidus.
-
Acute disseminated histiocytosis —a disease with prominent cutaneous, visceral, and bone marrow involvement occurring mainly in infants. This is referred to as Letterer-Siwe disease.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


