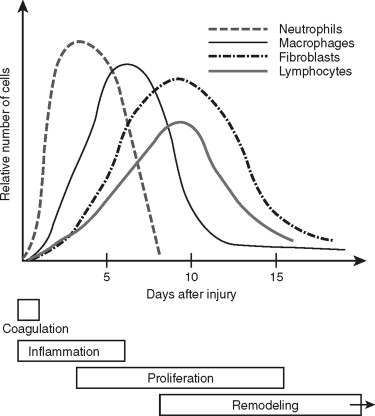
Traumatic wounds occur in a spectrum, from simple linear lip lacerations to complex panfacial fractures. The response to these tissue injuries is a complicated and dynamic process with four basic phases: coagulation, inflammation, proliferation, and remodeling ( Figure 2-1 ). The appearance of the different types into the wound suggests their function and importance during that phase of repair. The ultimate goal of wound repair is to return tissue function as quickly as possible. The rapid repair that is required for our survival still must provide good mechanical strength, eliminate pathogenic contamination, and inhibit excessive fluid loss and penetration. To achieve this end, the body replaces structurally complex tissue with fibrosis. Fibrosis satisfies most of the body’s requirements, but it is not ideal. For example, repaired skin tissue is less pliable and has reduced strength compared with uninjured tissue. Therefore, we must use our understanding of wound repair to minimize the amount of fibrosis and attain optimal results for our patients.
In this review of wound healing, I have focused on cutaneous and bone repair, since these are the two types of tissue we deal with most frequently in the trauma setting.
COAGULATION
Thrombus formation is the first key step in the wound repair process. Clot formation prevents excessive blood loss after mechanically induced trauma. Platelets initially activate after exposure to collagen in the perivascular tissues ( Figure 2-2 ). Von Willebrand factor mediates this interaction. Activated platelets release adenosine diphosphate (ADP) via exocytosis of dense granules and generate thromboxane A2. These factors, in turn, activate other platelets, thereby propagating surrounding platelet activation. The importance of ADP receptor activation of platelets is illustrated by drugs such as clopidogrel, which block the platelet receptor for ADP and inhibit thrombogenesis. The platelet changes shape and aggregate, forming a delicate plug that requires bolstering to be able to resist shear forces. A meshwork of cross-linked fibrin provides the structural support to the platelet plug.
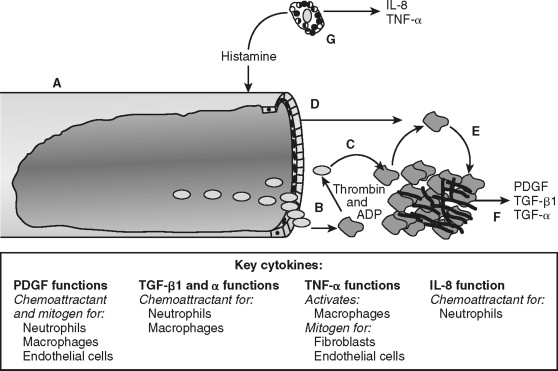
As platelets become activated, coagulation factors within blood are exposed to the surrounding tissue. Specifically, factor VII binds to tissue factor (its receptor) and initiates the coagulation cascade. The cascade proceeds as an ordered series of proteolytic reactions, which culminates in the generation of cross-linked fibrin. Activated platelets provide the phospholipid biolayer required for these enzymatic reactions. Ultimately, the tangled web of platelets and fibrin form a shear-resistant clot. The activation of coagulation and platelets links hemostasis with wound repair in addition to stopping the hemorrhage.
Establishment of hemostasis provides the initial environment necessary for wound repair to proceed. The clot serves as a temporary extracellular matrix that migrating cells use throughout the wound repair process. Activated clotting factor XII cleaves high-molecular-weight kininogen, which liberates bradykinin. This reaction occurs on the surface of platelets, neutrophils, and endothelial cells. Bradykinin mediates acute pain associated with inflammation directly by the interaction with bradykinin receptors found on sensory nerves. Bradykinin modulates pain indirectly by nociceptor sensitization and causes release of neuropeptides involved in nociception, including substance P. Thrombin generated during coagulation and released from platelets activates protease-activated receptor-1 (PAR-1) on the surface of endothelial cells (reviewed ). This signals endothelial cells to modulate surface selectins and integrins, which permit the rolling and eventual attachment of immune cells. The clot also serves as a reservoir of cytokines and growth factors, such as platelet-derived growth factor (PDGF) and transforming growth factor–beta (TGF-β) released from activated platelets. Thus, coagulation prevents excessive blood loss during trauma and initiates the inflammatory phase of wound repair at the site of injury.
INFLAMMATION
Mast cells participate in the initial phase of the inflammatory process. Classically, mast cells were simply thought to be involved in immediate hypersensitivity reactions to allergens and chronic inflammatory conditions. There is strong evidence that these cells also participate during wound repair, since mast cells are found in large numbers adjacent to epithelial and mucosal surfaces and release a myriad of inflammatory and cell-to-cell signaling mediators from stored vesicles. For example, activated mast cells release vesicles containing IL-8 and TNF-α, which recruit and activate neutrophils. Activated mast cells also release histamine. Histamine causes arteriolar vasodilation and venous constriction and increased capillary permeability, which contribute to the heat, erythema, and edema associated with inflammation. In animal wound–healing models, depletion of mast cells leads to aberrant wound repair. In these mice, full-thickness wound closure was slower and recruitment of neutrophils to the area was impaired. Histamine blockage in mice with normal numbers of mast cells produced abnormal wound repair similar to the mast cell deficient mice. This suggests that the source of the histamine required for normal wound repair is mast cells. In addition, mast cells are the ideal cell to initially detect and respond quickly to injury. They provide the signaling required to recruit neutrophils and modulate endothelial cell permeability.
Endothelial cells allow transmigration of immune cells from blood to the injured tissue ( Figure 2-3 ). This is an active process that requires both endothelial cell and immune cell participation. For example, PAR-1 receptor activation of endothelial cells leads to surface expression of selectins, which permits leukocyte rolling on the endothelial cell exterior. Leukocyte binding via selectins stimulates increased affinity to surface integrins, such as intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). This allows firm neutrophil attachment to endothelial cells. In addition, the signals received during this interaction are required for endothelial cell-to-cell junction separation. The mechanism of junction separation is not completely understood, but matrix metalloproteinases are involved. Matrix metalloproteinases are a family of enzymes that degrade extracellular matrix and cell surface proteins. Neutrophils migrate between endothelial cells to the injured area. Thus, endothelial cells have an active role in the inflammatory process by allowing the transmigration of immune cells.
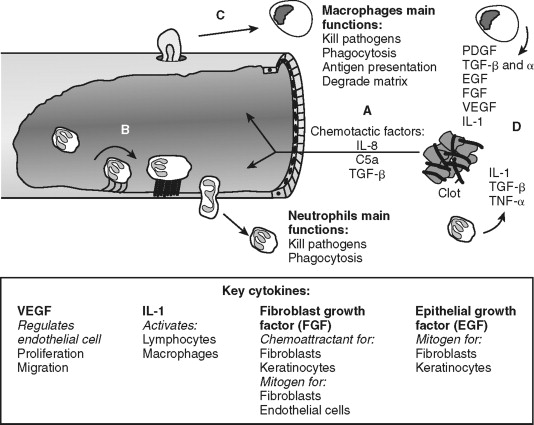
Neutrophils are the predominant cell type within the wound 24 hours after injury and function to kill bacteria and debride injured tissue. They are attracted to the area and activated by cytokines released from platelets, mast cells and by-products of the complement cascade. For example, complement factors C3a and C5 promote leukocyte chemotaxis and expression of ICAM-1. Activated neutrophils generate and release oxygen-free radicals from lysosomes. The oxygen-free radicals are antimicrobial but also cause tissue injury and neutrophil cell death; thus, their release is tightly controlled. Neutrophils debride the wound by removal of foreign debris and necrotic cells by phagocytosis. Neutrophils limit the inflammatory process by quickly eliminating microbes and tissue remnants. Prolonged inflammation caused by infection inhibits wound re-epithelialization. This delay in wound healing is not apparent in non-infected cutaneous or oral mucosal wounds in neutropenic animals suggesting the antimicrobial tissue debridement functions of neutrophil is essential for optimal wound healing. In contrast, collagen formation, wound closure, and strength are not significantly affected in neutropenic animals, suggesting that neutrophils are major participants in direct tissue wound repair. Neutrophil cell numbers decline dramatically 48 hours after injury as macrophages arrive and predominate in the wound.
Macrophages continue the effort of neutrophils and create the environment necessary to transition from the inflammatory to the proliferative phase of wound repair (see Figure 2-3 ). Blood monocytes arrive at the wound by transmigration through vessels in an analogous manner as neutrophils. Monocytes differentiate into active macrophages following interaction with the extracellular matrix, growth factors, and cytokines (see Figure 2-2 ). These activated macrophages produce critical cytokines, including IL-1 and TGF-β, that amplify inflammation. Macrophages also produce monocyte chemoattractant protein-1 (MCP-1), and macrophage-derived chemokines (MDC) that attracts lymphocytes to the wound. They direct the local lymphocyte response by becoming antigen-presenting cells. In addition, macrophages kill microbes directly by generating free radicals and hydrogen peroxide. Macrophages secrete matrix metalloproteinases, such as collagenase, that degrade the extracellular matrix in preparation for endothelial and fibroblast cell migration and proliferation within the wound. With all these functions, it is easy to surmise that macrophages are an essential cell type during wound healing. Indeed, monocyte-deficient animals demonstrate reduced wound strength, collagen production, and re-epithelialization rates.
INFLAMMATION
Mast cells participate in the initial phase of the inflammatory process. Classically, mast cells were simply thought to be involved in immediate hypersensitivity reactions to allergens and chronic inflammatory conditions. There is strong evidence that these cells also participate during wound repair, since mast cells are found in large numbers adjacent to epithelial and mucosal surfaces and release a myriad of inflammatory and cell-to-cell signaling mediators from stored vesicles. For example, activated mast cells release vesicles containing IL-8 and TNF-α, which recruit and activate neutrophils. Activated mast cells also release histamine. Histamine causes arteriolar vasodilation and venous constriction and increased capillary permeability, which contribute to the heat, erythema, and edema associated with inflammation. In animal wound–healing models, depletion of mast cells leads to aberrant wound repair. In these mice, full-thickness wound closure was slower and recruitment of neutrophils to the area was impaired. Histamine blockage in mice with normal numbers of mast cells produced abnormal wound repair similar to the mast cell deficient mice. This suggests that the source of the histamine required for normal wound repair is mast cells. In addition, mast cells are the ideal cell to initially detect and respond quickly to injury. They provide the signaling required to recruit neutrophils and modulate endothelial cell permeability.
Endothelial cells allow transmigration of immune cells from blood to the injured tissue ( Figure 2-3 ). This is an active process that requires both endothelial cell and immune cell participation. For example, PAR-1 receptor activation of endothelial cells leads to surface expression of selectins, which permits leukocyte rolling on the endothelial cell exterior. Leukocyte binding via selectins stimulates increased affinity to surface integrins, such as intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). This allows firm neutrophil attachment to endothelial cells. In addition, the signals received during this interaction are required for endothelial cell-to-cell junction separation. The mechanism of junction separation is not completely understood, but matrix metalloproteinases are involved. Matrix metalloproteinases are a family of enzymes that degrade extracellular matrix and cell surface proteins. Neutrophils migrate between endothelial cells to the injured area. Thus, endothelial cells have an active role in the inflammatory process by allowing the transmigration of immune cells.
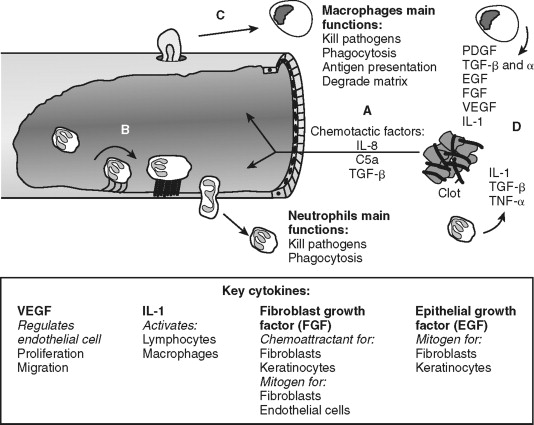
Neutrophils are the predominant cell type within the wound 24 hours after injury and function to kill bacteria and debride injured tissue. They are attracted to the area and activated by cytokines released from platelets, mast cells and by-products of the complement cascade. For example, complement factors C3a and C5 promote leukocyte chemotaxis and expression of ICAM-1. Activated neutrophils generate and release oxygen-free radicals from lysosomes. The oxygen-free radicals are antimicrobial but also cause tissue injury and neutrophil cell death; thus, their release is tightly controlled. Neutrophils debride the wound by removal of foreign debris and necrotic cells by phagocytosis. Neutrophils limit the inflammatory process by quickly eliminating microbes and tissue remnants. Prolonged inflammation caused by infection inhibits wound re-epithelialization. This delay in wound healing is not apparent in non-infected cutaneous or oral mucosal wounds in neutropenic animals suggesting the antimicrobial tissue debridement functions of neutrophil is essential for optimal wound healing. In contrast, collagen formation, wound closure, and strength are not significantly affected in neutropenic animals, suggesting that neutrophils are major participants in direct tissue wound repair. Neutrophil cell numbers decline dramatically 48 hours after injury as macrophages arrive and predominate in the wound.
Macrophages continue the effort of neutrophils and create the environment necessary to transition from the inflammatory to the proliferative phase of wound repair (see Figure 2-3 ). Blood monocytes arrive at the wound by transmigration through vessels in an analogous manner as neutrophils. Monocytes differentiate into active macrophages following interaction with the extracellular matrix, growth factors, and cytokines (see Figure 2-2 ). These activated macrophages produce critical cytokines, including IL-1 and TGF-β, that amplify inflammation. Macrophages also produce monocyte chemoattractant protein-1 (MCP-1), and macrophage-derived chemokines (MDC) that attracts lymphocytes to the wound. They direct the local lymphocyte response by becoming antigen-presenting cells. In addition, macrophages kill microbes directly by generating free radicals and hydrogen peroxide. Macrophages secrete matrix metalloproteinases, such as collagenase, that degrade the extracellular matrix in preparation for endothelial and fibroblast cell migration and proliferation within the wound. With all these functions, it is easy to surmise that macrophages are an essential cell type during wound healing. Indeed, monocyte-deficient animals demonstrate reduced wound strength, collagen production, and re-epithelialization rates.
PROLIFERATION
Formation of granulation tissue marks the initiation of the proliferative phase of wound healing 72 hours after injury. The granulation tissue is thought to provide the micro-environment required for wound contraction and re-epithelialization. It mainly consists of endothelial cells and fibroblasts. These cells gain access to the injured area after extracellular matrix degradation by matrix metalloproteinase secreted by macrophages (reviewed ). These cells migrate through the wound using the fibrin meshwork created during clot formation. Fibroblasts and endothelial cells are stimulated to proliferate and migrate by cytokines and growth factors secreted mainly by macrophages ( Figure 2-4
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


