Fig. 13.1 • Splancnocranium in a 5 week embryo: differentiation of Meckel’s cartilage.
Branchial arches, therefore, consist of a central mesodermal core externally covered by ectoderm and internally lined by endoderm; from the mesodermal central nucleus originate the typical components of each branchial arch: a cartilagineous bar, from which derive the skeletal structures, an artery, a muscular component and later the neural components. The internal mesenchymal layer is rich of neural crest cells, which will constitute the future head and neck region. Each branchial arch will give origin to a precise number of elements.
First branchial arch
Regarding the skeletal component, the first branchial or mandibular arch, has a fundamental importance. The first branchial arch is located between the stomodeum and the first pharyngeal pouch; it shall give origin to the bones of the two lower thirds of the face. Within the first pharyngeal arch two different processes may be isolated: the maxillary and the mandibular process, respectively in a more dorsal and in a more anterior position, located laterally and inferiorly to the stomodeum. From the maxillary process derive the structures that constitute the maxilla and the palate: the maxillary bone, the premaxillary bone, the zygomatic bone and the squamous part of the temporal bone.
Within the mandibular process is located the Meckel’s cartilage, which will disappear with development, with the exception of two small portions which will form the incus and the malleus.
The mandibular bone will form by ossification of the mesenchymal tissue of the Meckel’s cartilage itself (Fig. 13.2).
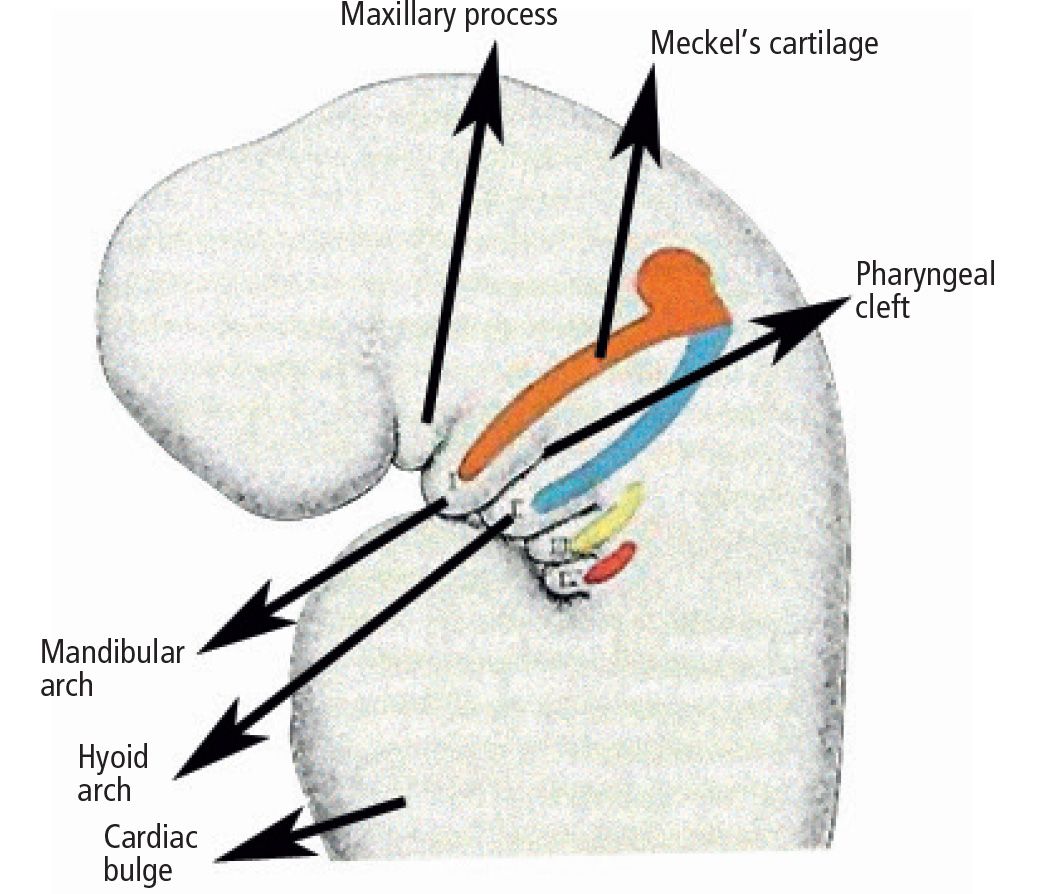
Fig. 13.2 • Splancnocranium of a 5 week embryo: Meckel’s cartilage differentiation.
From the mesoderm of the first arch also originate the muscular tissues which will constitute the muscles of mastication (masseter, temporalis, external and internal pterygoid), the mylohyoid muscle, the anterior belly the dygastric muscle, the tensor timpani muscle and the tensor veli palatini muscle. These muscles receive their nerve supply by the trigeminal nerve.
The mucosa and all the glands of the two anterior thirds of the tongue derive from the ectoderm and from the endoderm of the first branchial arch.
The second arch
The second pharyngeal arch will form the elements of the first part of the neck. The skeletal components comprise the styloid process of the temporal bone and the stapes, as well as the upper part of the hyoid bone (the body and the lesser cornu). The muscles which will originate are the stylohyoid muscle, stapedius muscle, the posterior belly of the dygastric muscle, the anterior and posterior auricular muscles and the muscles of facial expression. Innervation derives from the facial nerve.
The third arch
The third arch will give origin to the greater cornu of the hyoid bone and to the lower part of the body of the hyoid bone. The only muscle which originates from the third arch is the stylopharyngeous muscle. Innervation is given by the glossopharyngeal nerve.
The fourth arch
The fourth pharyngeal arch will form all the anatomical structures together with the sixth pharyngeal arch. Cartilagineous components of these arches fuse to form the thyroid cartilage, the cricoid cartilage, the arytenoid cartilage, the corniculate and cuneiform cartilages of the larynx. The muscular part that originates from the fourth arch involves the cricothyroid muscle, the levator veli palatini muscle and the constrictor muscles of the pharynx. Innervation is given by the superior laryngeal branch of the vagus nerve.
The sixth arch
The sixth arch will form the inferior portion of the thyroid cartilage, the cricoid cartilage and the arytenoids, cuneiform and corniculated cartilages. The intrinsic muscles of the larynx (superior and lateral crycoaritenoid muscles, arytenoid muscle and tyroarytenoid muscle). Innervation is given by the recurrent laryngeal branch of the vagal nerve.
The human embryo, furthermore, has five pairs of pharyngeal pouches.
The last of these, is atypical, and is often considered as part of the fourth. Various organs differentiate from the endoderm of these pouches:
- the first branchial pouch gives origin to the epithelial lining of the auditory tube, of the tympanic membrane and of the middle ear;
- the second branchial pouch gives origin to crypts of the palatine tonsils (tags of endodermal formations which are then invaded by B lymphocytes which then transform into lymphoid organs);
- the third branchial pouch: the solid dorsal bulbar portion of the third pouch differentiates into the parathyroid tissue, whereas the elongated ventral portion of the third pouch forms the first part of the thymus. The parathyroid tissue of the third pouch later forms in the adult the inferior parathyroid gland;
- the fourth branchial pouch: the ephitelium of the dorsal bulbar portion of this pouch forms the superior parathyroid gland;
- The fifth branchial pouch is generally considered as part of the fourth pouch. It gives origin to the last branchial body which is later incorporated in the thyroid gland. In the adult, the cells of the last branchial body give origin to the parafollicular cells or C cells of the thyroid, which produce calcitonin, a hormone which controls calcium level in the blood.
Anomalies within first and second branchial arch syndromes may therefore include several structures: the internal ear, the middle ear and the external ear, the condyles, the coronoid, the ramus and the body of the mandible, the masticatory muscles, the zygomatic bones, the inferior and lateral portion of the orbit and of the zygomatic arch, the temporomandibular joint, the parathyroid, the seventh and in some cases the third and the fourth and the fifth cranial nerve (Fig. 13.3).
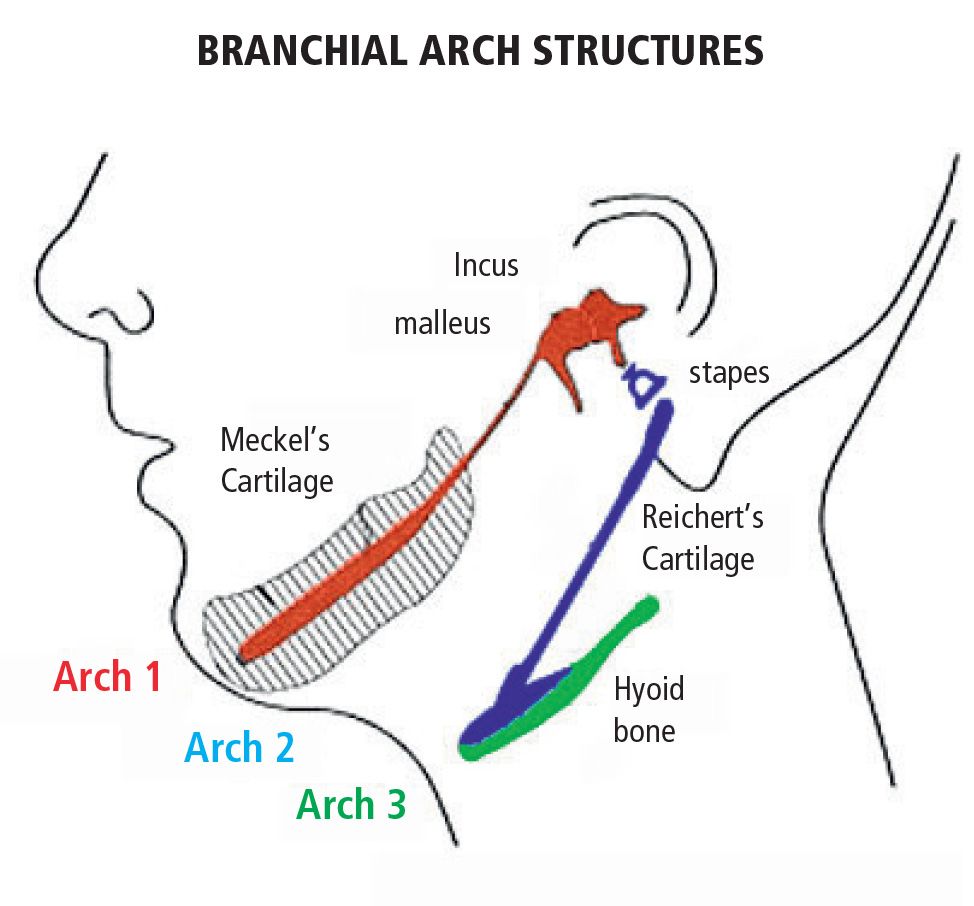
Fig. 13.3 • Derivatives of the I e II branchial arches.
In all these conditions the involvement is not limited exclusively to the face, but also cardiac, kidney, skeletal and central nervous system anomalies may be associated. The principal conditions are summarized in Table 13.1.
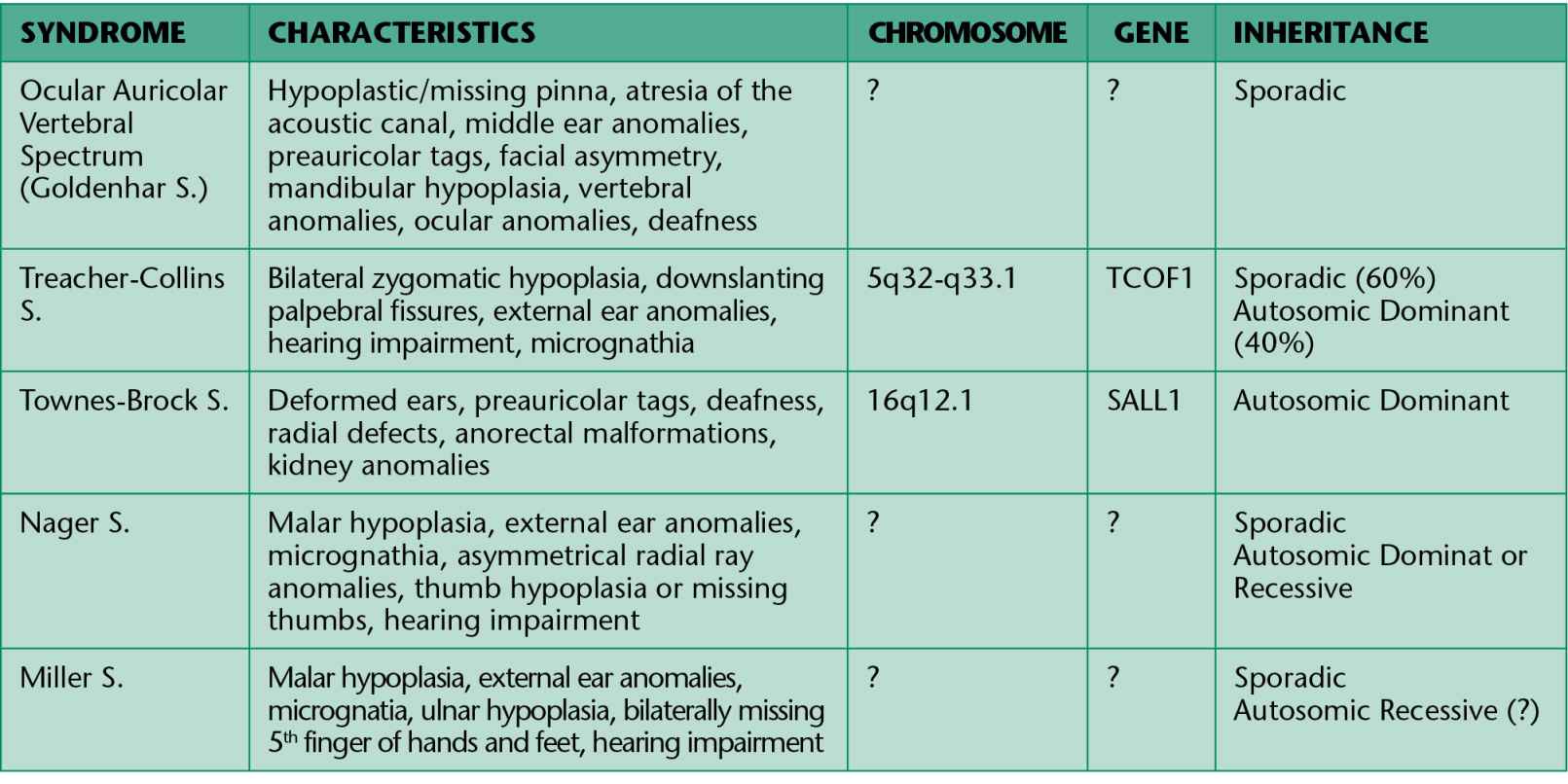
Table 13.1
Nowadays there is a tendency to group the clinical cases here described in a group of malformations called “LATERAL FACIAL MICROSOMIA” or “OTOMANDIBULAR DYSPLASIAS”.
We shall describe the two most common conditions relating to the anomaly of development of the first and second branchial arches.
TREACHER COLLINS SYNDROME
The Franceschetti-Klein syndrome or Treacher Collins syndrome is a genetically autosomic domi- nant disease with a penetrance of 90% and variable expressivity. Its incidence is around one over 50,000 newborn. This syndrome is associated to: auricular hypoplasia (77%), atresia of the external auditory canal (36%), anomalies of the ossicular chain, conductive deafness (40%); mandibular and zygomatic hypoplasia (80%), down-slanting palpebral fissures, inferior eyelid coloboma (69%), absence of the eyelashes on the lateral third of the inferior eyelid, mandibular hypoplasia (78%), palatal cleft (28%). Facial deformities are bilateral and symmetrical. Intelligence is generally normal. Given the narrowness of the upper respiratory tract there might be early respiratory distress. Hearing impairment should be evaluated as early as possible and prosthetic support should be precocious. Treatment is symptomatic with breathing assistance to correct respiratory distress. The number of new mutations is around 60%, but it’s probably lower according to recent molecular studies. It is generally due to a mutation of the gene TC 0F1. The gene is localized on the 5q32-q33.1 chromosome.
GOLDENHAR SYNDROME
Goldenhar syndrome, also known as ocular auricular vertebral dysplasia (or OAV), is a rare disease, characterized by a triad (generally unilateral) of craniofacial microsomia, ocular dermal cysts and anomalies of the vertebral column. The presence of auricular anomalies is absolutely essential to the diagnosis. The incidence of OAV is approximately 1/5.000-25.000 live births; it is more frequent in males. In the majority of cases it is sporadic; in 1-2% of the patients a dominant transmission is observed; some families have been described with a possible autosomic recessive transmission. Etiology is still unknown. At this point in time, the most probable hypothesis is that of a defect in the formation of the mesoderm or a defect in the interaction between the neural crest cells and the mesoderm. Many factors have been hypothesized in the development of the syndrome: some drugs (cocaine, thalidomide, retinoic acid, tamoxiphen), environmental factors (insecticides, herbicides) and maternal diabetes.
Ocular anomalies are present in about 50% of the cases; the most common ones are the dermoid and the epibulbar lipodermoid. They may not be seen on a CT scan or with a magnetic resonance imaging (MRI) as they are very similar to the orbital fat. Auricular defects are seen in 65% of cases and vary from preauricular tags, microtia, anotia and conductive deafness. Vertebral anomalies include vertebral agenesis, hemivertebrae, fusion of the ribs, kyphosis and scoliosis. Other typical characteristics are cardiovascular anomalies, genitourinary anomalies and pulmonary anomalies. Cardio respiratory distress in the first months of life is a common complication and threatening for the life of the patient. The syndrome may be associated to zygomatic bone hypoplasia, mandibular hypoplasia, maxillary hypoplasia, facial muscles hypoplasia and tongue anomalies, cleft lip and palate and central nervous system anomalies.
14. clinical features OF FIRST AND SECOND BRANCHIAL ARCH SYNDROMES
(M.C. Meazzini)
HEMIFACIAL MICROSOMIA (OTO-MANDIBULAR SYNDROME, OTO-MANDIBULAR DYSOSTOSIS), THE “OCULAR AURICULAR VERTEBRAL SPECTRUM”
This deformity is characterized by facial asymmetry, evident in 65% of the cases, but severe only in 20% of the cases, which is usually unilateral, even though bilateral cases exist in 10 to 33% of cases.
There is a mild predominance of the male subjects with a ratio between males and females of 3 to 2 and the right side is more affected, with a ratio between right side and left side of 3 to 2 (Burglen, 2001).
Hemifacial microsomia is characterized by a great variability in clinical manifestations.
Only derivatives of the first branchial arch, or all derivatives of the second branchial arches may be involved.
In the first case, the ramus, the mandibular condyle, the maxilla, the temporomandibular joint, the masticatory muscles, the oral commissure and the components of the ear which derive from the first branchial arch may be involved: the tragus, the base of the Helix, the incus and the malleus, and the tensor timpani muscle (Fig. 14.1).
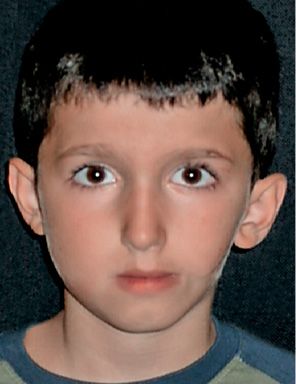
Fig. 14.1 • Frontal photograph of a patient affected by a mild form of HFM in which the structures originating from the first branchial arches are more involved: the ramus and mandibular condyle, the maxillary bone, the masticatory muscles. A macrostomia was present on the affected side. A preauricular tag was present, but the auricle is only mildly displaced and anteriorly rotated.
In those cases, where the structures originating from the second branchial arch are involved as well, the facial nerve may be affected, totally or partially, the segments of the ears which derive from the second arch: the stapes, the Helix, the anti-Helix, the anti-tragus, the lobule (Bettega, 2001; Burglen, 2001) (Fig. 14.2).
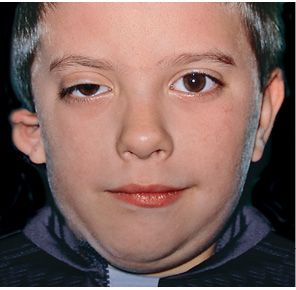
Fig. 14.2 • Frontal photograph of a patient affected by a more severe form of HFM in which the structures originating from the first branchial arch as well as those from the second branchial arch are involved. The facial nerve is affected and the auricle is severely deformed.
The variety of the phenotypic expression is such, that we may see a deformity which is so slight, that there might only be fibro-cartilaginous pre-auricular tags (anterior to the tragus), or we may see patients with complete absence of the ear, or a very hypoplastic ear positioned inferiorly and anteriorly relative to the contralateral, with absence of the ear canal, missing temporo-mandibular joint and masticatory muscles and a severe deformity of the hemimandible (Brusati, 1988).
In the most striking cases the mandibular ramus together with the coronoyd process, is short, or in some cases, absent.
The mandibular angle on the affected side is more cranial and medial relative to the contralateral; the chin is deviated to the affected side and the mandibular lower border is higher than the contralateral.
The occlusal plane is usually oblique, the maxilla is shorter on the affected side secondary to the intrinsic hypo-development, both sagittal and vertical, of the maxilla (first branchial arch), which is very evident at the molar level.
The occlusion is a class II occlusion on the affected side and the lower midline is deviated towards the affected side. Often the mandibular arch, is particularly short, and presents a severe crowding and molar hypo eruption, especially on the affected side, with overeruption at the lower incisal area (Bettega, 2001).
The temporo-mandibular joint may be affected with different levels of severity. Its severity has guided the classification of Pruzansky for the mandibular deformity in hemifacial microsomia in 1969 and later modified by Murray in 1987 and in 1988 by Kaban (Pruzansky, 1969; Murray, 1984; Kaban, 1988) (Figg. 14.3a-d).
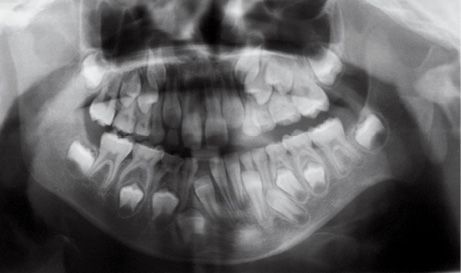
Fig. 14.3a • Condylar shape in a patient affected by an HFM with mandibular deformity Type I (Pruzanski, 1969). The condyle and ramus are normal but smaller.
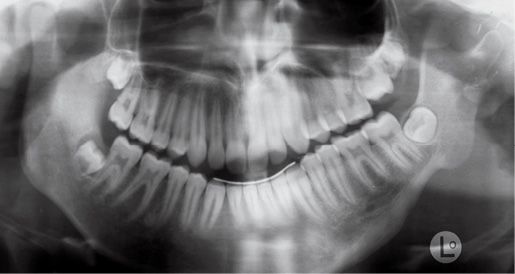
Fig. 14.3b • Condylar shape in a patient affected by an HFM with mandibular deformity Type IIa (Pruzanski, 1969; Kaban, 1988). The condyle is deformed in terms of shape and size, but the relationship with the glenoid fossa is maintained.
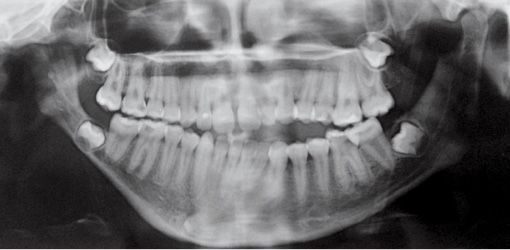
Fig. 14.3c • Condylar shape in a patient affected by an HFM with mandibular deformity Type IIb (Pruzanski, 1969; Kaban, 1988). The condyle is severely deformed in terms of shape and size. The TMJ is rudimentary and anteriorly and medially dislocated.
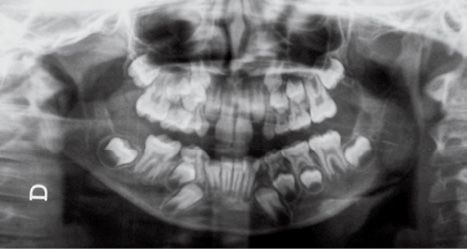
Fig. 14.3d • Condylar shape in a patient affected by an HFM with mandibular deformity Type III (Pruzanski, 1969). The condyle, the coronoid process and the TMJ and the proximal portion of the ramus are absent.
PRUZANSKI’s CLASSIFICATION
Type I: the condyle is normal but smaller;
Type IIa: the condyle is deformed in terms of shape and dimensions, but the relationship between the glenoid fossa and the condyle is maintained and the fossa on the affected side has a position within the temporal bone which is similar to that of the contralateral side. Articular function is substantially maintained;
Type IIb: the condyle is severely deformed with a rudimentary temporomandibular joint, anteriorly and medially dislocated and displaced;
Type III: condyle, coronoid, temporomandibular joint (both the disc and the fossa) and the proximal portion of the ramus are absent.
SAT CLASSIFICATION
David in 1987 has introduced a classification which reminds the general principles of the cancer classification TNM. This classification was called SAT, where “S” means the skeletal involvement, “A” means the auricular condition and “T” means the the soft tissue involvement (David, 1987). T1, minimal defect and no involvement of the cranial nerves; T2, moderate anomalies; T3 major hypoplasia generally associated to cranial nerves involvement, of the salivary glands, and of the masticatory muscles.
OMENS CLASSIFICATION
Vento in 1991 has proposed a system of classification called OMENS for hemifacial microsomia: the acronym stands for “O”, the involvement of the ocular region, ”M” mandibular, “E” auricular (ear), “N” central nervous system and “S” stands for the soft tissue involvement. To each of these deformities, points from 1 to 3 are given considering their severity (Vento, 1991).
At the ocular level the presence of epibulbar malformations (dermoids, lipodermoids) is present in 35 to 50% of the cases. Their presence may influence vision, but their removal may be very dangerous. A moderate dystopia of the external canthus may be present and a moderate lowering of the whole orbit. It is possible, though not consistent, to find a microphthalmus or an anophthalmus. The zygoma is slightly hypoplastic. In the more severe forms, the zygomatic arch is absent and its anterior remnant is inferiorly directed. There is often a deviation of the nose towards the affected side and the cheek is generally hypoplastic because of the missing or hypoplastic musculature and because of the deformities of the zygomatic region, the ramus, the mandible and the temporomandibular joint.
The muscular defects reduce the articular motion: opening, protrusion, and lateral movements are limited. When the patient opens the mouth, the mandible deviates towards the affected side, because of the absence of an adequate leverage on the affected condyle in its corresponding glenoid fossa. The difficulty of bringing the mandible towards the affected side is also related to both skeletal and muscular associated anomalies. Often times the temporal muscles and the masseter muscles are fused together and constitute a common muscular group, the temporo-masseter sling (Marsh, 1989).
Mimic musculature maybe normal or have severe defects, depending on how involved the facial nerve is. Some authors believe it to be involved in more than 45% of the cases (McCarthy, 1990) (Fig. 14.4).
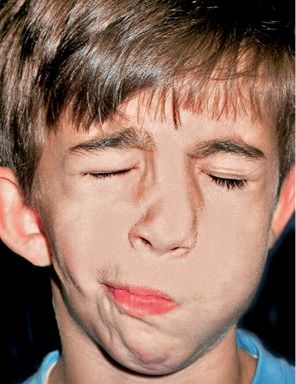
Fig. 14.4 • Frontal photograph of a patient affected by HFM in which the structures originating from the first branchial arch as well as those from the second branchial arch are involved. The facial nerve is affected with a secondary defect in the mimic musculature.
The involvement of other cranial nerves is possible, the cochlear nerve, responsible for a sensorineural hearing impairment, needs to be checked for, and distinguished from a conductive type of hearing disorder, which is much more frequent, given the hypoplasia of the auricular structures (Bettega, 2001). In 42% of the patients vertebral and rib anomalies may be present (vertebral fusion, hemi-vertebrae, especially cervical, bifid spinal cord), in 28% of the cases there might be cardiac involvement (interventricular defects) or in 10% of the cases respiratory and gastrointestinal or genitourinary anomalies might be present (Gorlin, 2001).
According to what Gorlin proposed in 1990, geneticists have grouped under the denomination “Ocular auricular vertebral spectrum” the isolated asymmetrical forms, unilateral or bilateral of oto-mandibular dysostosis and Goldenhar syndrome or ocular auricular dysplasia. Goldenhar syndrome, which represents 10% of the cases, has the same clinical aspect and the aetiology of hemifacial microsomia, but it may be distinguished for the frequent asymmetrical bilateral involvement of the defects, the less frequent involvement of the temporo-mandibular joint and ramus and the more frequent association to other deformities such as the vertebral anomalies. The ocular involvement is also typical with epibulbar dermoids, or coloboma of the upper eyelid (Gorlin, 2001).
Treacher Collins syndrome, Franceschetti syndrome or mandibulo facial dysostosis
It is a congenital syndrome characterized by bilateral deformities of the orbit, eyelids and maxillo-mandibular complex, masticatory muscles and ears (Fig. 14.5) (Burglen, 2001).
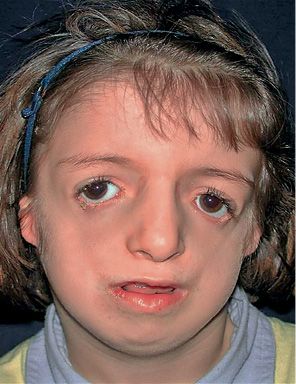
Fig. 14.5 • Frontal photograph of a patient affected by Treacher Collins Syndrome. Note the downslanting palpebral fissures, the severely hypoplastic zygomatic arches, and the coloboma of the lower eyelids.
According to Tessier’s classification on facial clefts, Treacher Collins Syndrome is characterized by the presence of a cleft n. 6 (maxillo-zygomatic), a cleft n. 7 (temporo-zygomatic) and a cleft n. 8 (fronto-zygomatic).
The frontal process of the zygomatic bone is hypoplastic or missing, the palpebral fissures have an outward downward slanting.
In 75% of the patients there is a coloboma of the inferior eyelid with absence of eyelashes on the medial portion.
The nose is prominent with an obtuse naso-frontal angle.
A choanal atresia is frquent and correlated to the severe vertical hypodevelopment of the posterior portion of the maxillary bone.
The chin is retruded, the mandibular angle is obtuse.
Ears may be missing or abnormal.
The hearing bones of the ossicular chain may be abnormal and in 55% of the patients there is a bilateral conductive hearing impairment.
Maxillary bone and mandibular ramus have a severe verical hypoplasia.
The masseter muscle is inserted, given the absence of the zygoma, into the temporal bone aponeurosis.
Cleft palate is present in more than 30% of the patients (Gorlin, 2001).
There may be a class I occlusion with a severe clockwise rotation of the occlusal plane.
An open bite may be present, and a bilateral cross bite with constriction of the maxillary arch is common.
The mandibular plane presents a severe downward rotation (Brusati e Sesenna, 1988). (Fig. 14.6)
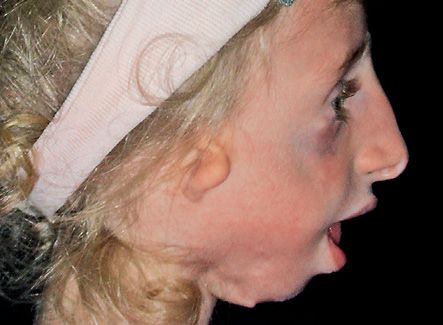
Fig. 14.6 • Facial profile view of a patient affected by Treacher Collins Syndrome. Note the severe mandibular retrusion.
Dental crowding, tooth rotations, dental anomalies, such as congenitally missing teeth, and impacted teeth are frequent.
Acro- facial dysostosis: Nager syndrome, Miller syndrome
Acro-facial dysostosis is a heterogeneous group of disorders where craniofacial anomalies are combined to developmental disorders of the limbs.
NAGER SYNDROME
The most well-known pre-axial form (referring to the lateral/radial aspect of the upper arm) is called Nager syndrome (Fig. 14.7).
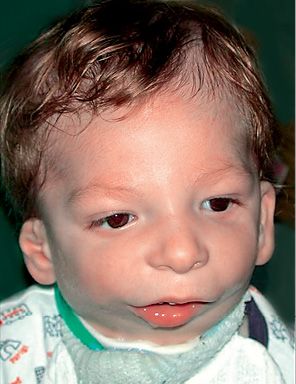
Fig. 14.7 • Frontal photograph of a patient affected by Nager Syndrome. Note the mildly downslanting palpebral fissures, the bilateral asymmetrical deformity of the auricles.
It was first recognized by Nager in 1948, and presents asymmetrical deformity of the limbs, such as absence of the radius ( about 100% of the patients) and radio-ulnar synostosis, absence or hypoplasia of the thumbs and loss of the flexion-extension ability of the thumbs, sindactyly, clinodactily, frequent proximal radio-ulnar synostosis. Lower limbs may, although not consistently, be affected (Bergland, 2001). The face is characterised by an anti-mongoloid inclination of the palpebral rims (100% of the patients) with the presence of a coloboma of the eyelid in 50% of the patients, malar region hypoplasia, external ear anomalies (in 80% of the patients) very similar to those seen in Treacher Collins syndrome with low set ears and ear hypoplasia. Often there is bilateral hearing impairment. There is no angle between the neck and the chin. It is often associated to cleft palate. The forehead may be prominent. Bilateral mandibular hypoplasia with severe micrognathia and retrogenia which may be associated to severe respiratory distress (Burglen, 2001) (Fig. 14.8) is usually present. Cardiovascular anomalies and genitourinary anomalies may be associated (Gorlin, 2001).
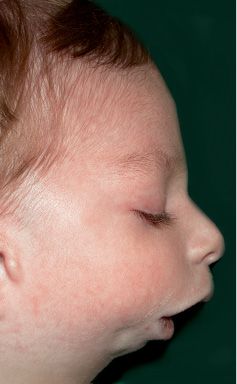
Fig. 14.8 • Facial profile view of a patient affected by Nager Syndrome. Note the very severe mandibular retrusion correlated to the severe respiratory distress.
MILLER SYNDROME
The most common post-axial syndrome is Miller syndrome. Miller syndrome is a rare autosomal recessive condition that mainly affects the development of the face and limbs. The severity of this disorder varies among affected individuals. Miller syndrome is a rare disorder; it is estimated to affect less than 1 in 1 million newborns. At least 30 cases have been reported in the medical literature.
Children with Miller syndrome are characterized by malar hypoplasia and micrognathia. They often have a cleft palate and/or cleft lip. Respiratory distress related to the micrognathia is common. Patients with Miller syndrome have downward slanting eyelids, ectropion and lower eyelid coloboma. Hypotia and conductive hearing loss are present.
The most common limb problem is the absence of the fifth fingers and toes. Affected individuals may also have syndactyly. Less commonly, affected individuals have abnormalities of the heart, kidneys, genitalia, or gastrointestinal tract.
Mutations in the DHODH (dihydroorotate dehydrogenase) gene cause Miller syndrome.
REFERENCES – ClinicAL FEATURES
Bettega G., Morand B., Lebeau J., Raphaël B. Les altérations morphologiques au cours des syndromes oto-mandibulaires. Ann. Chir. Plast. Esthét. 46: 495, 2001.
Brusati R., Sesenna E. Chirurgia delle deformità mascellari. Milano, Masson, 1988.
Burglen L., Soupre V., Diner P.A., Gonzalès M., Vazquez M.P. Dysplasis oto-mandibulaires: Génétique et nomenclature des formes syndromiques. Ann. Chir. Plast. Esthét. 46: 400, 2001.
David D.J., Mahatumarat C., Cooter R.D. Hemifacialmicrosomia: a multisystem classification. Plast and Reconstr Surg. 80: 525, 1987.
Gorlin R., Cohen M.M. Jr, Hennekam R.C.M. Syndromes of the Head and Neck. Oxford University Press 2001.
Kaban L.B., Moses M.H., Mulliken J.B., Surgical correction of hemifacial microsomia. Plast and Reconstr Surg. 82: 9. 1988.
Marsh J.L., Baca D., Vannier M.W. Facial musculoskeletal asymmetry in hemifacial microsomia. Cleft Palate J. 26: 292-302, 1989.
McCarthy J.G. Plastic Surgery vol. 4 Cleft Lip and Palate and Craniofacial Anomalies. Saunders 1990.
Murray J.E., Kaban L.B., Mulliken J.B. Analysis and treatment of hemifacial microsomia. Plast Reconstr Surg. 74: 186-99, 1984.
Pruzansky S. Not all dwarfed mandibles are alike. Birth Defects. 1: 120, 1969.
Vento A.R. et al. The O.M.E.N.S. classification of hemifacial microsomia. Cleft Palate Craniofac. J. 28: 68-76, 1991.
Vigneron J., Stricker M., Vert P., Rousselot J.M., Levy M. Postaxial acrofacial dysostosis (Miller) syndrome: a new case. J Med Genet. 28: 636-8, 1991.
15. CRANIOFACIAL GROWTH IN FIRST AND SECOND BRANCHIAL ARCH SYNDROMES
(M.C. Meazzini)
GROWTH IN UNOPERATED HEMIFACIAL MICROSOMIA PATIENTS
One of the most controversial topics in the literature, regards the progressive nature of the clinical asymmetry during growth of a patient affected by hemifacial microsomia. Converse (1973), Murray (1984), Kaban (1998) and Kearns (2000) suggest that the congenital alteration of the functional matrix is such (muscles, bone, periosteum) that the anomaly worsens during growth. The progressiveness of the asymmetry would thereby dictate the need for surgical intervention as early as possible. Polley et al., on the other hand, following the growth from 6 months of age till the end of craniofacial growth of 26 HFM non operated patients on serial postero-anterior cephalometric X-rays, have demonstrated that the affected side of the mandible continues to grow with a rate that maintains the vertical ratio between the affected and the non affected side (Polley, 1997). The same was demonstrated in a later study on panoramic X-rays of non operated patients from 5 years till the completion of growth (Meaz-zini, 2011a). This means that the facial proportion of the patient affected by hemifacial microsomia is maintained throughout the whole process of craniofacial growth (Figg. 15.1a-d).
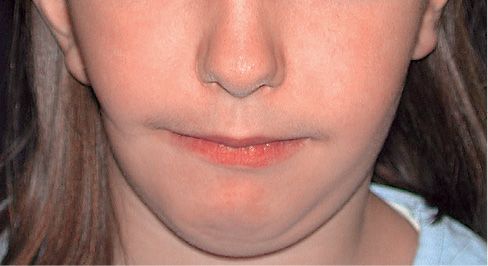
Fig. 15.1a • Frontal facial photograph of a patient affected by hemifacial microsomia at 4 years of age.
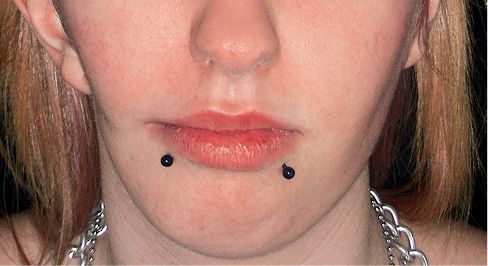
Fig. 15.1b • Frontal facial photograph of a patient affected by hemifacial microsomia at 15 years of age, never subjected to any surgery or orthopaedic treatment. Note that the degree of asymmetry did not change with growth.
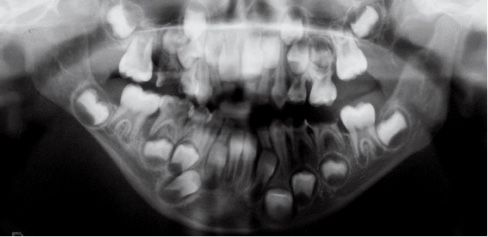
Fig. 15.1c • Panoramic Radiograph of a patient affected by Type IIb hemifacial microsomia at 4 years of age.
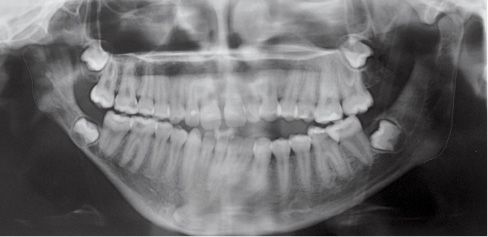
Fig. 15.1d • Panoramic Radiograph of a patient affected by Type IIb hemifacial microsomia at 15 years of age, never subjected to any surgery or orthopaedic treatment. Note that the ratio affected ramus/non affected ramus did not change with growth.
As described by Mommaerts and Nagy (2002), much confusion exists between the rate of growth, meaning the yearly amount of growth, and the ratio between the affected and the non affected side. According to Rune (2004) and Polley (1997), the growth of the affected side maintains the same proportion relative to the non affected side so that the degree of asymmetry remains relatively constant during the development of the child. This is also true for the maxillary bone (Meazzini, 1997), which maintains the same inclination throughout the years. This may be explained by the fact that the affected side grows, but not as much as the non affected side. Therefore, the fact that the proportions are maintained, means, unmistakably, that the affected side during time grows less than the non affected side.
GROWTH OF HEMIFACIAL MICROSOMIA PATIENTS AFTER COSTOCHONDRAL GRAFTING
Growth after costochondral graft (described in detail in chapter 16) is known to be highly unpredictable. In many cases there is a variable recurrence of the asymmetry with time. Less frequently the costochondral graft may grow excessively. In some cases ankylosis of the graft may occur. In a study conducted at San Paolo Hospital (Milano), we compared the growth after costochondral graft of patients affected by hemifacial microsomia (congenital asymmetries) and patients affected by unilateral temporomandibular joint ankylosis (acquired asymmetry). The results of this study have shown that in the patients with the acquired asymmetry there is more commonly a balanced growth or, at times, an excessive growth on the grafted side, whereas, in the congenital forms, the growth of the graft, although clinically significant, in most patients is unable to maintain the facial symmetry throughout growth (Meazzini et al 2011b) (Figg. 15.2a-f).
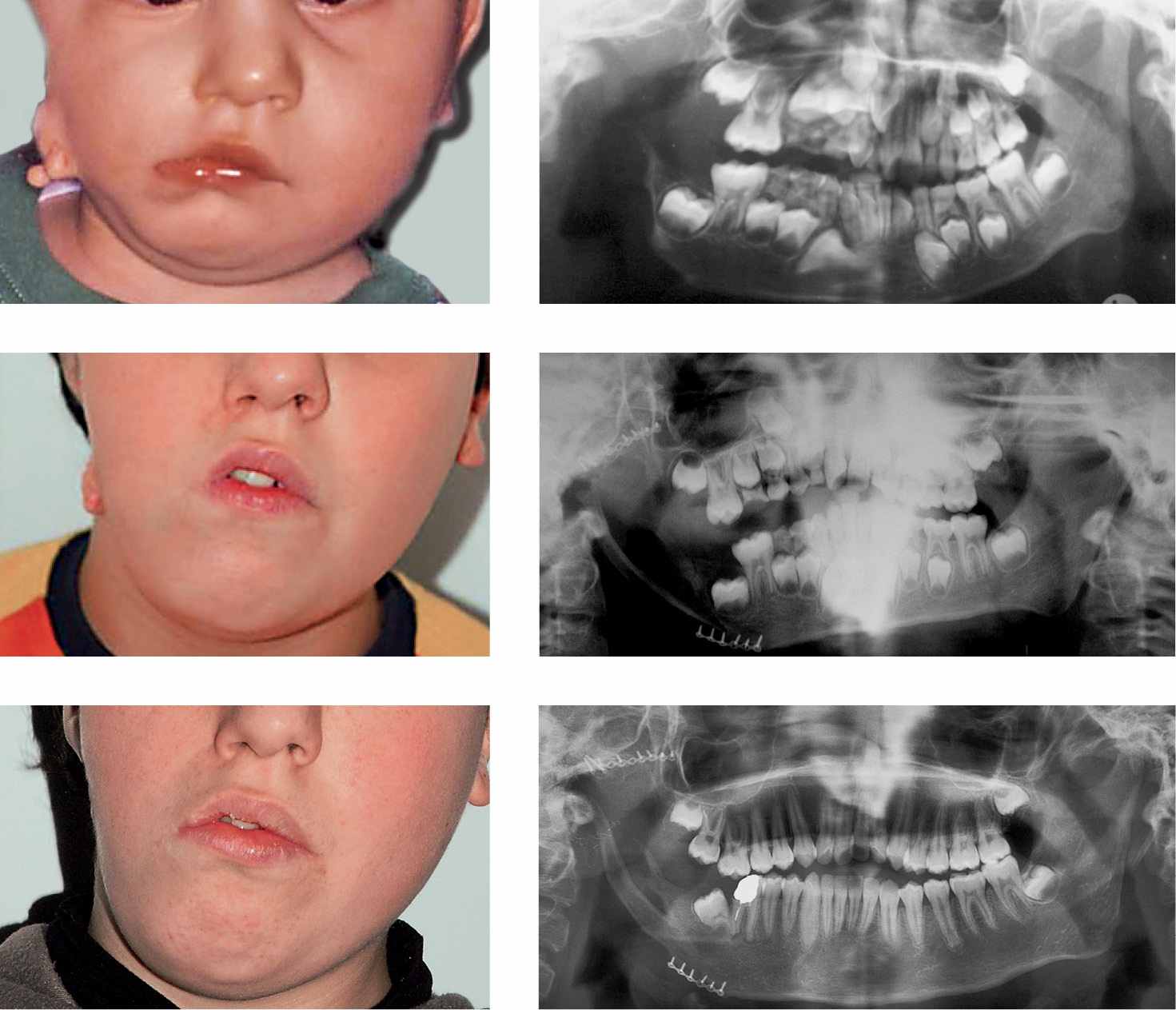
Figg. 15.2a-f • a. Frontal facial photograph of a patient affected by hemifacial microsomia Pruzanski III (congenital asymmetry) before surgery. b. Panoramic Radiograph of the patient before costochondral graft. c. Panoramic Radiograph of the patient after costochondral graft (Prof. Brusati). d. Frontal facial photograph of a patient affected by hemifacial microsomia Pruzanski III (congenital asymmetry) 7 years after costochondral graft (Prof. Brusati): note the asymmetrical growth. e-f. Panoramic Radiograph of the patient 7 years after costochondral graft. The growth of the graft does not proceed with the same rate of the non affected side.
According to many authors early surgery is indicated to prevent maxillary deformities, which are believed to be secondary to a primary mandibular deformity (Mulliken and Kaban 1987; Kaban,1998). Nevertheless, Padwa suggests to postpone costochondral grafting at least till early adolescence, in order to obtain a more stable result (Padwa, 1998). Interestingly, in a study conducted in the department of Maxillo-facial surgery at the San Gerardo Hospital (Monza) we have evaluated a sample of patients affected by HFM who had been subjected to unilateral mandibular distraction osteogenesis. In these patients the whole “deforming” effect of the mandible on the maxillary complex was therefore “removed” early by distraction osteogenesis. The study was a 5 year follow-up, conducted on a very homogeneous sample in terms of age at the time of surgery (average age 5.6 years +/-0.6) and in terms of the initial severity of the mandibular hypoplasia (type I and type II according to Pruzansky’s classification). The patients, have been monitored with lateral and postero-anterior cephalometric X-rays and with panoramic X-rays immediately before treatment, right at the end of the distraction lengthening and every year of follow-up stop the longest follow up for all the patients was on average 5.8+2-0.4 years. The results of this study showed that immediately post distraction, at one year, at three years and at five years post distraction the asymmetry in the infra orbital plane and in the nasal floor (maxillary skeletal base) did not change. On the contrary the Occlusal plane inclination (dento-alveolar component of the maxilla) was reduced of an average of 9° between the preoperative and the end of the distraction period. During the first year post operatively this reduction was maintained. There was a gradual return towards the original Occlusal plane inclination with an average increase in the subsequent five years of 4° (about 40% of the improvement obtained with the distraction process). These data seem to demonstrate that thanks to distraction osteogenesis a significant dento-alveolar remodeling of the maxilla was possible, but no true skeletal modifications of the base of the maxilla were obtained. Therefore, as already mentioned, the potential for adaptation by the dento-alveolar portion of the maxillary bone, as every orthodontists knows, is extremely high, but the defect in the skeletal base remains. This result seems to reduce the conviction that an early correction of the hypoplastic mandible might be an actual benefit on the skeletal maxillary- zygomatic growth and in preventing secondary deformities. The deformity of the maxillary base, after 5 years of age, does not improve after distraction osteogenesis, and only the dento-alveolar part of the maxilla remodels after mandibular correction. Therefore, it is reasonable to believe that the maxilary deformities are in reality primary deformities associated to the first branchial arch defect in HFM, and this is the reason why there is no spontaneous correction of the maxillary skeletal base once the mandibular interference is eliminated. Only the dento-alveolar component of the maxilla is secondarily remodeled (Meazzini, 2005).
GROWTH AFTER DISTRACTION OSTEOGENESIS OF THE MANDIBLE
In the study which we have just mentioned, it was also shown that, in terms of ratio between the affected ramus and the non affected ramus, at the end of distraction there was a perfect symmetry. Nevertheless, 5 years post-distraction 70% of the correction obtained with DO was lost (Meazzini, 2005). In a more recent long term study with a follow up between 10 and 13 years post-DO, it was demonstrated that all of the correction in terms of ratio was lost, and the patients presented the same proportions they had pre-DO (Meazzini, 2011). In figure 15.3 a follow-up 13 years post distraction osteogenesis in a patient affected by hemifacial microsomia Type II operated at 5.4 years of age is shown (Fig. 15.3).
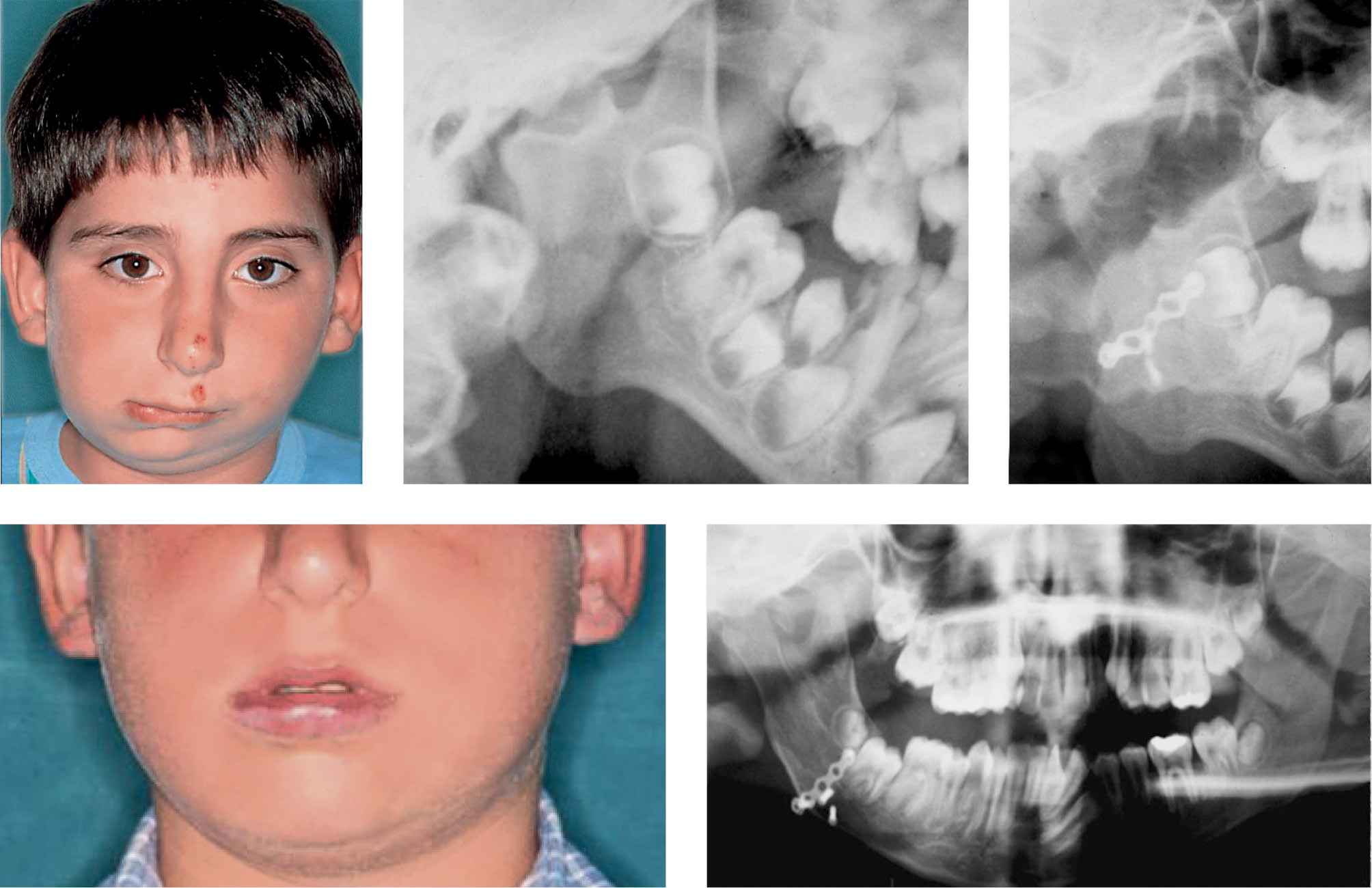
Figg. 15.3a-e • a. Frontal facial photograph of a patient affected by unilateral TMJ ankylosis (acquired asymmetry) before costochondral graft. b. Panoramic Radiograph of the patient before costochondral graft. c. Panoramic Radiograph of the patient after costochondral graft (Prof. Brusati). d. Frontal facial photograph of the patient affected 8 years after costochondral graft (Prof. Brusati): note the symmetrical growth. e. Panoramic Radiograph of the patient 8 years after costochondral graft. The growth of the graft proceeds with the same rate of the non affected side.
In an early study on a very small sample of six patients affected by hemifacial microsomia a reccurrence of the mandibular morphology registered during the first year after distraction osteogenesis had been extremely high, ranging from 23 to 52% (Meazzini, 1997). At the time we were hoping that the association of functional jaw orthopaedics, might help to reduce this recurrence of the phenotype. In the study mentioned above (Meazzini, 2005) it was noted that during the first year post-DO the “return” (relapse) had been much less (chapter 17). It was therefore hypothesized, that the pre-DO and post-DO orthopaedic treatment that these patients had been subjected to, might have helped to maintain a better stability of the correction during the first year post distraction. In order to demonstrate if the combined orthopaedic-distraction treatment had any real advantages, not in the short term, but in the long term, we needed to compare two groups of patients, treated consecutively with two different protocols: one group of patients had no orthopaedic preparation before or after DO (only occlusal adjustments), while the second group of patients had at least one year of orthopaedic treatment prior to DO and one year post-DO. The results of this study demonstrated that orthopaedic treatment may slow down, but does not stop, the process of “return” to the original pattern of asymmetry and, again, the beneficial influence of orthopaedic treatment expresses itself more at the dento-alveolar level then at a skeletal level (Meazzini, 2008).
Although the term “return” is usually not as clinically clear as the term “relapse”, these authors have preferred it because, what is truly happening in these patients, is not an actual relapse. When we talk about relapse we think about loss of the regenerate, whereas, in this case, the return to the original pattern is due to a differential growth which is directly linked to the congenital pathologic pattern of growth. Stability of the regenerate is usually measured during the first six months to one year after DO. The literature is generally in agreement that there is very little actual relapse (loss of regenerate) after DO. The controversy is concentrated it’s long-term effects. DO guarantees the achievement of perfect symmetry in the short-term. In the long term, mandibular morphology is remodeled through a process of re-expression of the syndrome specific pattern of growth.
The contradiction in hemifacial microsomia was originated by the belief that the growth of hemifacial microsomia patients was of a progressive nature. The literature seems now oriented towards the conclusion that hemifacial microsomia patients, whether operated or not, present, at the end of growth, an asymmetry which has a very similar proportion to the initial disproportion (Hollier, 1999; Kusnoto, 1999; Gosain, 2001; Swennen, 2001; Mommaerts and Nagy, 2002; Meaz-zini, 2005 and 2011a).
Very similar observations were made in patients with bilateral mandibular hypoplasia affected by Nager syndrome, Treacher Collins syndrome or other very severe bilateral congenital micrognathia. In a follow-up longer than 1.5 years post-DO, patients affected by these syndromes have shown the progressive re-expression of their syndrome specific mandibular deformity (Stelnicky, 2002). There is therefore a need to realize that DO is just a tool, extremely efficient in its ability to modify the skeletal structure of young patients, but not their genetic code. This knowledge forces us to select patients with great care and to be honest and clear with families regarding long term prognosis, making it very clear, that the hope that an early correction through DO will be a definitive procedure, is extremely unrealistic (Fig. 15.4).
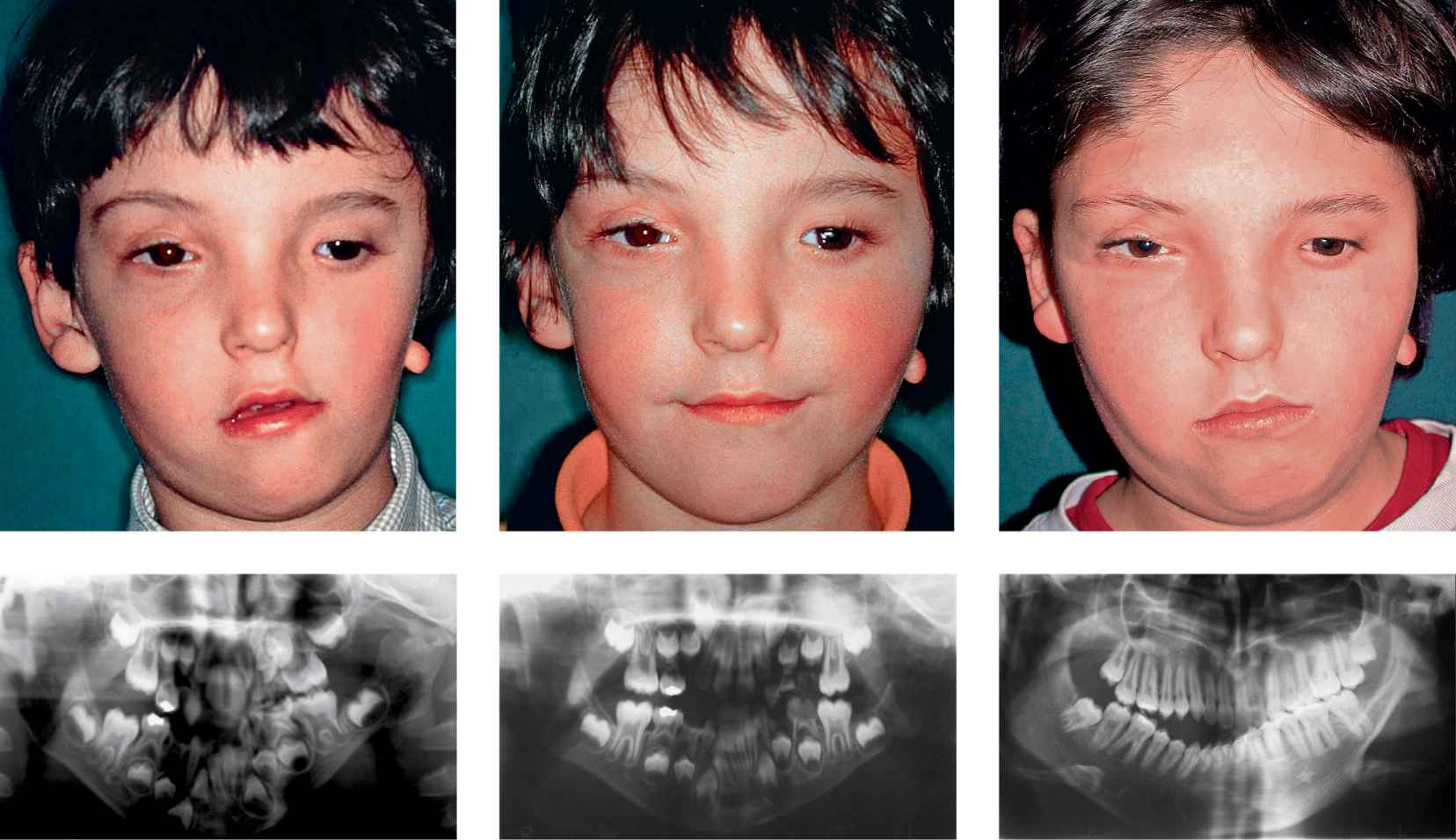
Figg. 15.4a-e • a. Frontal facial photograph of a patient affected by hemifacial microsomia Pruzanski IIb before distraction osteogenesis. b. Panoramic Radiograph of the patient affected by hemifacial microsomia Pruzanski IIb before distraction osteogenesis. c. Frontal facial photograph of the patient post distraction (Dr. Mazzoleni): note the good symmetry achieved. d. Panoramic Radiograph of the patient 1 year post distraction osteogenesis, note the tendency towards an asymmetrical growth. d. Frontal facial photograph of the patient 13 years post distraction: note the asymmetrical growth. e. Panoramic Radiograph of the patient 13 years post distraction: the growth of the distracted side does not proceed with the same rate of the non affected side and the ratio affected/non affected side has returned to the pre-DO ratio.
REFERENCES – GROWTH
Converse J.M., Coccaro P.J., Becker M., Wood-Smith D. On hemifacial microsomia. The first and second branchial arch syndrome. Plast Reconstr Surg. 51: 268-79. 1973.
Gosain A.K. Distraction osteogenesis of the craniofacial skeleton. Plast Reconstr Surg. 107: 278, 2001.
Hollier L.H., Gosain A., Stelnecki E., Longaker M., McCarthy J.G. Symposium, Craniofacial Distraction Osteogenesis. The Journal of Craniofacial Surgery. 10: 268, 1999.
Hollier L.H., Kim J.H., Grayson B., McCarthy J.G. Mandibular growth after distraction in patients under 48 months of age. Plast Reconstr Surg. 103: 1361-70, 1999.
Kaban L.B., Padwa B.L., Mulliken J.B. Surgical correction of mandibular hypoplasia in hemifacial microsomia: the case for treatment in early childhood. J Oral Maxillofac Surg. 56: 628-38, 1998.
Kearns G.J., Padwa B.L., Mulliken J.B., Kaban L.B. Progression of facial asymmetry in hemifacial microsomia. Plastic and reconstructive surgery. 2: 492, 2000.
Kusnoto B., Figueroa A.A., Polley J.W. A longitudinal Three-dimensional evaluation of the growth pattern in Hemifacial Microsomia treated by mandibular distraction osteogenesis: A preliminary report. The Journal of Craniofacial Surgery. 10: 480, 1999.
Meazzini M.C., Figueroa A.A., Polley J.W. Maxillary changes in patients with Hemifacial microsomia after mandibular distraction osteogenesis. In: Diner P.A., Vasquez M.P. (Eds.), Proceedings of the 1st International Congress on Cranial and Facial Bone Distraction Processes. Monduzzi, Italy: Bologna, pag 115-119, 1997.
Meazzini M.C., Mazzoleni F., Bozzetti A., Caronni E. Orthopaedic treatment prior to mandibular distraction osteogenesis in growing patients with hemofacial microsomia. In: Diner P.A., Vasquez M.P. (Eds.), International Proceedings of the 2nd International Congress on Cranial and Facial Bone Distraction Processes. Monduzzi, Italy: Bologna, pag 81-86, 1999.
Meazzini M.C., Mazzoleni F., Canzi G., Bozzetti A. Mandibular distraction osteogenesis in hemifacial microsomia: long-term follow-up. J Craniomaxillofac Surg. 33: 370-6, 2005.
Meazzini MC, Mazzoleni F, Bozzetti A, Brusati R. Comparison of mandibular vertical growth in hemifacial microsomia patients treated with early distraction or not treated: Follow up till the completion of growth. J Craniomaxillofac Surg. 2011 Mar 29. [Epub ahead of print]
Meazzini MC., Mazzoleni F., Bozzetti A., Brusati R. Does functional appliance treatment truly improve stability of mandibular vertical distraction osteogenesis in hemifacial microsomia? J Craniomaxillofac Surg. 2008 Oct; 36(7): 384-9.
Mommaerts M.Y., Nagy K. Is early osteodistraction a solution for the ascending ramus compartment in hemifacial microsomia? A literature study. J Craniomaxillofac Surg. 30: 201-7, 2002.
Mommaerts MY, Nagy K. Is early osteodistraction a solution for the ascending ramus compartment in hemifacial microsomia? A literature study. J Craniomaxillofac Surg. 2002 Aug; 30(4): 201-7.
Mulliken J.B., Kaban L.B. Analysis and treatment of hemifacial microsomia in childhood. Clin Plast Surg. 14: 91-100, 1987.
Murray J.E., Kaban L.B., Mulliken J.B. Analysis and treatment of hemifacial microsomia. Plast Reconstr Surg. 74: 186-99, 1984.
Padwa P.O., Mulliken J.B., Kaban L. Midfacial growth afterchostochondral graft reconstruction of the mandibular ramus in hemifacial microsomia. Journal of Oral and Maxillofacial Surgery. 56: 122, 1998.
Polley J., Figueroa A.A., Liou E.J., Cohen M. Longitudinal analysis of mandibular asymmetry in hemifacial microsomia. Plast Reconstr Surg. 99: 328, 1997b.
Stelnicki E.J., Lin W.Y., Lee C., Grayson B.H., McCarthy J.G. Long-Term outcome study of bilateral mandibular distraction: a comparison of Treacher Collins and Nager Syndrome to Other types of micrognathia. Plast Reconstr Surg. 109: 1819, 2002.
Swennen G., Schliephake H., Dempf R., Schierle H., Malevez C. Craniofacial distraction osteogenesis: a review of the literature. Part 1: clinical studies. International Journal of Oral and Maxillofacial Surgery. 30: 89, 2001.
16. SURGICAL TREATMENT OF I AND II BRANCHIAL ARCH SYNDROMES
(F. Mazzoleni)
INTRODUCTION
The following chapter is meant to address the surgical treatment planning of craniofacial malformations, due to an aberrant development of those structures originating from the I and II branchial arch and the nasal placode (Hemifacial Microsomia or Otomandibular, Goldenhar, Franceschetti, Nager Syndromes etc.), both in growing patients and in adults.
The different syndromes of I and II branchial arches share many common features and differ from each other depending on the involvement modality of some craniofacial districts and other parts of the body. It’s important to distinguish those forms that are mainly unilateral from bilateral ones, according to the aim of this chapter and thus to the treatment modality.
Hemifacial Microsomia and Goldenhar Syndrome usually appear with an unilateral hypoplasia, while just 10-15% of cases are bilateral. In Franceschetti syndrome there’s a highly variable but mainly bilateral hypoplasia, while Nager Syndrome and Pierre Robin Sequence are exclusively bilateral 1-8.
The phenotypic expression of syndromes of I and II branchial arch is highly variable, depending both on the amount of involved sutures and the severity of the hypoplasia of each anatomical structure.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


