Fig. 6.1
Fibrillin individual domains. (a) Detailed structure of the fibrillin cbEGF13–14 pair. The amino acids are numbered from N- to C-terminus. The bound calcium ions (one per cbEFG domain) are indicated as black circles close to the N-terminus of each domain. The cysteine cross-links (three per cbEFG domain) are indicated by zigzag lines. (b) Fibrillin domain structure. The 43 cbEGF domains (blue) are numbered. There are four additional more complete epidermal growth factor (EGF) domains at the N-terminal region (gray). There are nine TB domains, but only seven are linker domains (green ovals). The TB1 domain is linked to a separate proline-rich domain (dark blue) and TB3 (linked to cbEGF10) is modified (green diamond). The TB linker domains are a potential source of flexibility, through their interaction with their flanking cbEGF domains. The interbead segment, TB4-6 and intervening cbEGF domains (orange segment at the foot of Fig. 4.17b) are the compressed, normal form that expands under tension by releasing Ca2+ to give the 70-nm beaded form (a – From Fig. 3 in Whiteman P. et al. (1998 Apr 3) “A Gly –> Ser change causes defective folding in vitro of calcium-binding epidermal growth factor-like domains from factor IX and fibrillin-1.” J. Biol. Chem. 273(14):7807–7813. b – Reprinted with minor changes from Arnaout MA (2004) “The structural basis of elasticity in fibrillin-based microfibrils.” Structure 12(4):734–736; Copyright 2004, with permission from Elsevier)
Fibrillin forms head-to-tail polymers that progressively fold at defined sites between successive molecules (Fig. 6.2). Initial internal folding at the head-tail junction (red/yellow) produces a 150-nm periodicity. Further folding at the proline-rich region (dark green) produces an approximately 100-nm bead periodicity and creates cross-links (not shown) between the folded regions. These cross-links are mediated by the enzyme transglutaminase, which replaces the terminal amide of glutamine with the ε-amino group of an adjacent lysine residue, creating an isopepide, intermolecular cross-link (Fig. 6.3). Transglutaminase also acts on various other proteins, most notably fibrinogen (Sect. 11.3.4). Transglutaminase requires calcium ions for activity and it attaches to folded (beaded) regions (light blue, egg-shaped region in Fig. 6.2) along with other proteins.
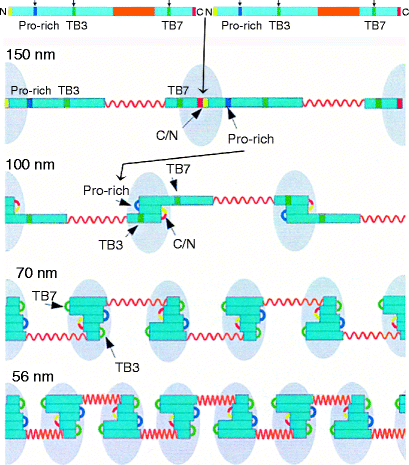
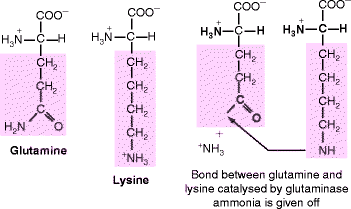
Fig. 6.2
The hinged model of fibrillin elasticity. Fibrillin forms dimers by covalent N- and C-terminal binding and this permits folding initiation to the 150-nm periodicity. As folding then proceeds to the 100-nm periodicity, glutaminase attaches and forms glutamine-lysine intra-molecular cross-links between the folded segments (blue). As folding proceeds further, the fibrillin polypeptide dimer bends at the TB3 and TB7 regions and additional proteins attach to this region. The N and C termini are in yellow and red, respectively. The interbead region between TB4 and TB6 is elastic. Its β-sheet structure loses calcium ions under tension, allowing disulfide-bonded cysteine residues to move by rotating away from each other (shown in orange). Numbers on the left indicate the periodicity. Details of folding are described in the text (Reprinted with minor changes from Structure, Vol 12 (4), Arnaout, MA, The structural basis of elasticity in fibrillin-based microfibrils, pp. 734–736; Copyright 2004, with permission from Elsevier)
Fig. 6.3
The transglutaminase reaction. Folding brings the glutamine and lysine residues into close proximity where bound glutaminase replaces the glutamine amide group by linking it to the ε-amino group of lysine. The amide is given off as ammonia
The extended central regions of the fibrillin polymer (orange in Fig. 6.2) remain free of associated proteins. Further intramolecular folding at the TB7- and TB3-cbEGF linker regions result in a ∼70-nm bead periodicity which corresponds to a “stretched” form. The interbead segment (cbEGF domains from TB4 to TB-6; orange in Fig. 6.2) spontaneously bind calcium ions, which compresses them further to give the observed 56-nm beaded periodicity, “relaxed” form. Stretching occurs within this segment and is reversible. On stretching, fibrillin periodicity increases from 56 to 70 nm due to the interbead segment dissociating bound calcium ions. When the stretching force is released, calcium ions can bind again and this pulls the segment back to the 56-nm “relaxed” form. The folded beaded region normally stays intact, but severe stretching that partially unfolds the beaded regions to above 100 nm prevents the beads from returning to the “relaxed” form (overstretched). Along with elastin (Sect. 6.2.1), fibrillin is an important component of ligaments (Sect. 3.1.3). If a ligament is overstretched it is the fibrillin microfibrils that are damaged and take a long time to be repaired.
Fibrillin-2 has an amino acid sequence that is 68% identical to fibrillin-1 and is coexpressed with fibrillin-1 in many tissues early in mammalian development. It forms head-to-tail fibrillin1/2 alternating heterodimers that resemble fibrillin-1 homodimers shown at the top of Fig. 6.3. During mammalian development, some tissues express fibrillin-2 without fibrillin-1 and fibrillin-2 homodimers may assemble by a mechanism that does not involve fibrillin-1 but perhaps utilizing fibrillin-3, a third member of the fibrillin family. Fibrillin-2 binds to the precursor of elastin during development and forms stronger elastic fibers than fibrillin-1. Fibrillin-3 is a minor component whose functions are uncertain.
Mutations of fibrillin-1 and -2 disrupt elastic tissue scaffolding, particularly in the aorta, eyes, and skin. An especially obvious effect of certain fibrillin-1 and -2 mutations is an overgrowth of the long bones of the body, resulting in long limbs and a tall stature (Marfan’s syndrome). In addition, there are major changes to the face, oral cavity, and teeth, most notably, a highly arched palate, crowded incompletely developed (hypoplastic) teeth and deformities of the roots. These changes may all stem from those fibrillin mutations that interfere with a normal interaction between TGF-β and BMP on the fibrillin surface. Mutations of stromal proteins that affect the teeth are listed in Table 7.1. It should be clear that fibrillin is absent from teeth and bones, which are essentially calcified type I collagen.
Table 6.1
Some common proteo-glycosaminoglycans (proteo-GAGs)
|
Proteoglycan
|
Core protein mol wt
|
Type of GAG chains
|
No. of chains
|
Location
|
Functions
|
|---|---|---|---|---|---|
|
Aggrecan
|
210,000
|
Chondroitin and keratan sulfate
|
∼130
|
Cartilage
|
Mechanical support; large aggregates with hyaluronan
|
|
Betaglycan
|
36,000
|
Chondroitin or dermatan sulfate
|
1
|
Cell surface and matrix
|
Binds TGF-beta
|
|
Decorin
|
40,000
|
Chondroitin or dermatan sulfate
|
1
|
All connective tissues
|
Binds type I collagen fibrils and TGF-beta
|
|
Perlecan
|
600,000
|
Heparan sulfate
|
2–15
|
Basal laminae
|
Structural and filtering function in basal lamina
|
|
Syndecan
|
32,000
|
Chondroitin and heparan sulfate
|
1–3
|
Epithelial cell surface
|
Cell adhesion; binds fibroblast and other growth factors
|
Summary
Fibrillin is the major component of beaded microfilaments possessing elasticity. It is secreted as a 150 nm polypeptide possessing almost 50 calcium-binding domains surrounded by flexible domains that allow folding. The calcium-binding domains are homologous to epidermal growth factor and the flexible domains to proteins that bind transforming growth factor-β. Fibrillin is secreted as covalently connected, head-to-tail dimers. The N- to C-terminal regions are central to the folding that provides the beaded appearance. Folding is stabilized by transglutaminase cross-linking between glutamine and lysine residues and various small proteins that bind to the folded (beaded) fibrillin. The central region of each molecule is free of associated proteins and consists of partially folded calcium-binding domains that stretch, expanding the bead periodicity from 56 nm to 70 nm. There are three homologous fibrillin molecules (fibrillin-1 through 3). Disruption of elastic tissue scaffolding due to mutations in fibrillin-1 and -2 affect the aorta, eyes, and skin. Some fibrillin mutations also cause abnormal bone growth (Marfan’s syndrome) perhaps due to an uncontrolled activation of latent TGF-β attached to microfibrils together with any bone morphogenic protein family member except BMP-1.
6.1.1 Elastin
Animals placed on a copper-deficient diet exhibit a decreased content of elastic fibers and suffer aneurysms of the aorta. This observation suggested an impaired cross-linking of elastin, and led to the isolation of a soluble precursor, tropoelastin (∼72,000 kDa), from the aortas of the copper-deficient animals. Elastin is encoded by a single gene with an exceptionally high intron/exon ratio. It is mainly expressed by fibroblasts and chondroblasts. Sequencing the tropoelastin polypeptide identified alternating short hydrophilic and hydrophobic domains. The hydrophilic domains are rich in lysine (K) with adjacent alanine (A) or proline (P) residues that become cross-linked (KA and KP domains). The hydrophobic domains are rich in valine (V), proline (P), and glycine (G), often as VPGVG or VGGVG repeats with or without alanine (A) and are ultimately responsible for the elasticity (Fig. 6.4a).
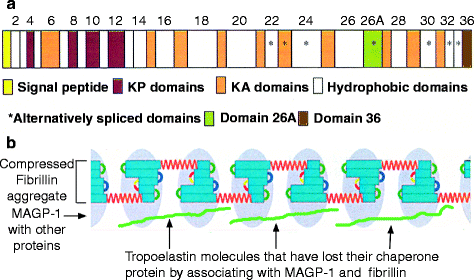
Fig. 6.4
Human tropoelastin domains. Each domain corresponds to an exon, which is irregular in size; the gene has a ratio of intron to exon DNA of about 20:1, one of the largest known. This ratio is only about 8:1 in fibrillar collagens. The signal peptide is cleaved in the endoplasmic reticulum, leaving tropoelastin. (a) Elastin has hydrophilic and hydrophobic domains. The lysine-rich KA and KP domains (colored) are hydrophilic and involved in cross-linking. They are interspersed within mostly longer hydrophobic regions (white). Asterisks indicate domains that may be included or spliced out (alternative splicing), permitting minor differences in elastin structure for different tissues. The C-terminal domain (domain 36) is essential for the interaction of tropoelastin with fibrillin to form elastic fibers (illustrated in b). Domain 26 is critical for the succeeding step, coacervation, whereby the fibrillin–tropoelastin aggregates form filamentous structures that then cross-link and develop into elastic fibers; recombinant tropoelastin lacking in domain 26 neither coacervates nor cross-links. Domain 26A is an exceptionally hydrophilic region that is usually spliced out. (b) Association of fibrillin with elastin. Fibrillin-associated protein MAGP-1 (light blue ovals) binds to elastin (green) by displacing the chaperone (a – Reprinted from Mithieux S, Weiss AS. (2005) “Elastin.” Adv. Prot. Chem. 70:437–461; with permission from Elsevier. b –Original figure derived by adding a representation of elastin to Fig. 6.2)
After tropoelastin is synthesized into the rough endoplasmic reticulum, its signal peptide is removed by a protease. The hydrophobic domains in the molecule bind to a 67-kDa chaperone, preventing self-aggregation and additional proteolysis. [Chaperones are proteins that mediate the folding of proteins and sometimes stabilize conformations that promote binding to other proteins.] The hydrophilic, C-terminal domain is a KP domain which does not bind the chaperone. This KP domain mediates the attachment of tropoelastin, mostly to fibrillin-2 microfibrils with bound microfibril-associated glycoprotein-1, (MAGP-1; Fig. 6.4b). The positively charged lysine residues of tropoelastin interact with the negatively charged fibrillin, forcing release of the chaperone and exposing the tropoelastin hydrophobic domains to the aqueous environment. Exposure of one such domain two-thirds of the way to the tropoelastin C-terminus causes the microfibril-bound tropoelastin molecules to repel water by coaggregating (coacervation). Coacervation causes the attached microfibrils to enclose the tropoelastin molecules, limiting their aqueous exposure. Coacervation is essential for the tropoelastin molecules to align for cross-linking. Later in life, the lack of expression of fibrillin-2 makes repaired elastic fibers weaker than those laid down during development.
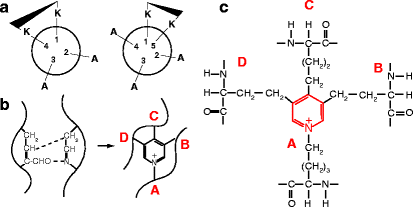
Cross-linking of the tropoelastin molecules within the tropoelastin–fibrillin aggregates is mediated by lysyl oxidase, the same enzyme responsible for cross-linking collagen fibers. In the KA domains, lysine residues are typically found in clusters of two or three amino acids, separated by two or three alanine residues. These regions are proposed to be α-helical with 3.6 residues per turn of helix, which has the effect of positioning two lysine side-chains on the same side of the helix, facilitating the formation of desmosine cross-links (Fig. 6.5). In the KP domains, the lysine pairs are flanked by prolines and bulky hydrophobic amino acids. Desmosine and isodesmosines have not been found in association with KP domains, probably due to the steric constraints imposed by the prolines. The loss of many positively charged lysine residues following cross-linking makes elastin fibers among the most insoluble proteins in the body and much less soluble than tropoelastin. It is not clear exactly how lysyl oxidase accesses the KA and KP domains at the center of the tropoelastin–fibrillin aggregates.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


