Fig. 9.1 • Locations of the mutations of the genes FGFR1, FGFR2 and FGFR3, related to CFS.
The vast majority of the mutations create aminoacidic substitutions, which usually induce the modification of the structure of the protein.
Functional studies have shown that all known mutations bring to a functional gain of the receptor protein, which in turn leads to an increased affinity of the bond to the ligands (as observed for the mutation Ser252Trp, which is responsible of 70% of the cases of Apert syndrome), or to the loss of specificity of the bond to the ligand (again in the Apert syndrome, it is observed in the mutation P253R).
The gene TWIST codes for a transcription factor, which is inserted in the same developmental path of the FGFRs receptors and acts immediately before these. Mutations of this gene lead to a loss of function of the protein and some functional studies have shown that a loss of function of the TWIST leads to an activation of the FGFR receptors (this leads to a functional gain the FGFR s).
Regarding the correlation between genotype and phenotype (between the mutation observed and the clinical presentation) the most significant, is the one observed in the Apert syndrome, where two mutations (Ser252Trp and P253R), induce aminoacidic substitutions of the two adjacent aminoacids, which are accountable for 99% of the cases. A larger incidence of cleft palate is observed in the first mutation and of severe syndactily in the second mutation (Emery et al, 2002).
In the other syndromes the correlations between phenotype and genotype are more difficult. The molecular data have shown that some craniosynostosis, which until now have been considered clinically different, may show identical mutations or be allelic diseases (different mutations of the same gene).
In table 9.1 we have summarized of the clinical and molecular data of the most common craniosynostosis.
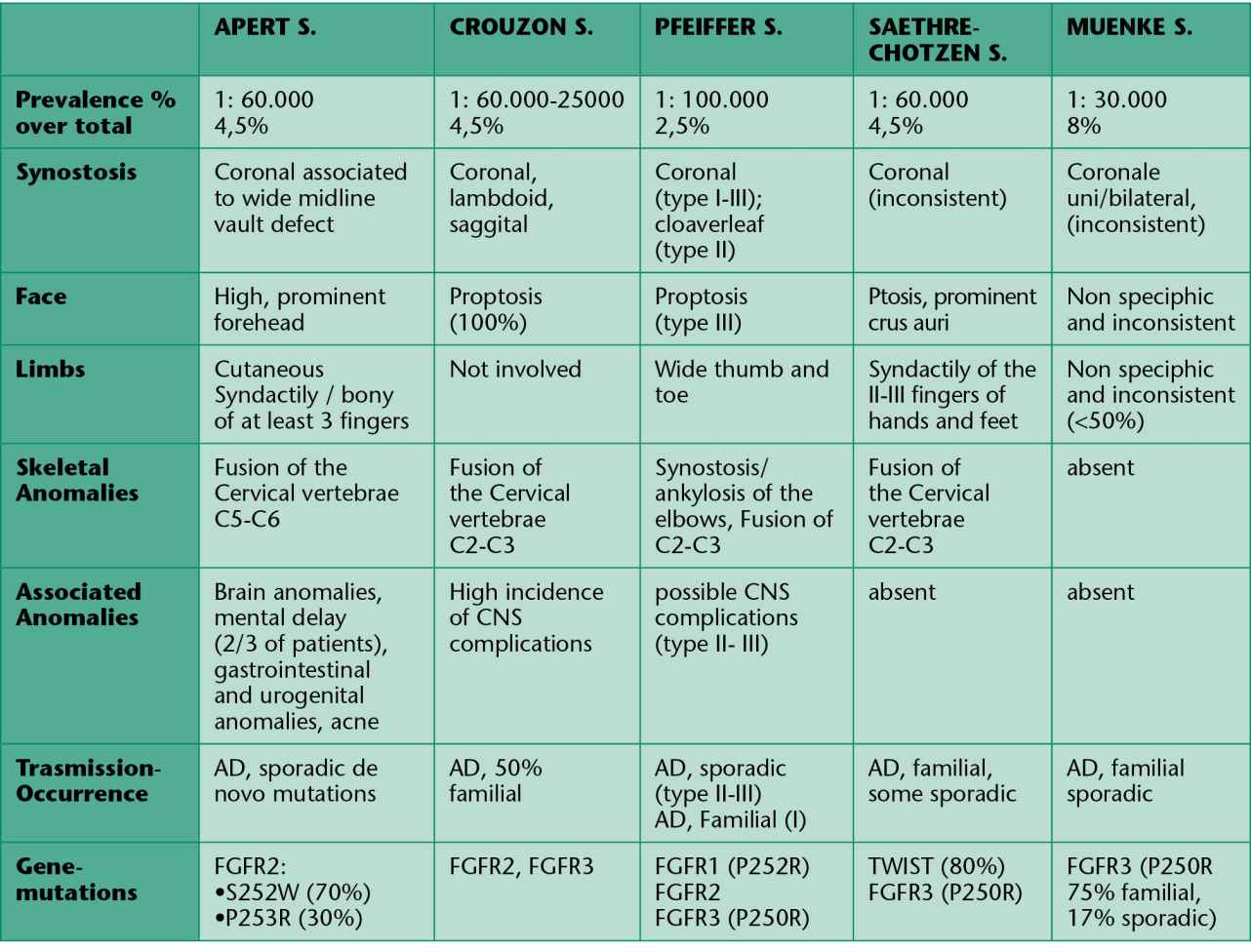
Tab. 9.1 • Clinical and genetic characteristics of some syndromic craniosynostosis.
REFERENCES – GENETICS
Cohen MM Junior MacLean RE. Craniosinostosis II ed. Oxford University Press 2002.
Emery DL, Connor JM, Pyeritz RE, Korf BR. Rimoin Emery and Rimoin’s Principles and Practice of Medical genetics Fourth edition. Churchill Livingstone 2002.
Cohen MM Jr. Perspectives on craniosynostosis: sutural biology, some well-known syndromes, and some unusual syndromes. J Craniofac Surg. 2009 Mar; 20 Suppl 1: 646-51.
Passos-Bueno MR, Serti Eacute AE, Jehee FS, Fanganiello R, Yeh E. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations Front Oral Biol. 2008; 12: 107-43.
10. EMBRYOLOGICAL DEVELOPMENT AND GROWTH IN CRANIOFACIAL SYNOSTOSIS AND clinical features
(M.C. Meazzini)
SHORT REVIEW OF EMBRYOLOGY OF THE NEUROCRANIUM AND OF THE CRANIAL BASE NEEDED TO BETTER UNDERSTAND CRANIOFACIAL SYNOSTOSIS
In the embryo, approximately during the fifth week, along the base of the skull, begins the formation of the chondrocranium, a structure formed by the confluence of more than one cartilaginous growth centre.
This cartilagineous cranial base acts as an embryological framework, which already presents the notches and foramens typical of the future bony cranial base.
On each side of this primitive chondrocranium begin to take shape cartilaginous formations, such as the parietal lamina, the temporal ala and the orbital ala, which have a fundamental role in the construction of the neurocranium.
Ossification at the level of the cranial base is mostly endochondral, and proceeds on the base of this cartilagineous structure.
As the ossification process proceeds, the various skeletal parts are not separated by fibrous tissue, as in sutures, but by cartilaginous tissue, which forms the spheno-occipital, the inter-sphenoidal and the spheno-ethmoidal syncondrosis.
These three syncondrosis of the cranial base are remnants of the primitive cranial base.
Of these, the most important in terms of growth, is the spheno-occipital, which controls the lengthening of the medial part of the cranial base and does not ossify during the whole period of infantile growth, while the cerebral hemispheres continue to grow.
At around seven years of age it stops being active and bony fusion starts and eventually ends at around 20 years of age (Roche, 1975).
In the cranial vault, on the other hand, the pattern of ossification is membranous and it creates skeletal subunits separated between each other by fibrous tissue, which secondarily form the actual cranial sutures (Enlow, 1975).
SHORT REVIEW OF CRANIOFACIAL GROWTH IN NORMAL CHILDREN NEEDED TO BETTER UNDERSTAND GROWTH IN CRANIOFACIAL SYNOSTOSIS
We may distinguish maxillary growth in prenatal and postnatal growth. Until the beginning of the deciduous dentition the premaxillary region grows entirely by apposition in an anterior direction, thereafter, a pattern of maxillary growth by posterior apposition and anterior resorption begins.
This change defines the passage between fetal growth and actual postnatal growth (Enlow, 1975).
Craniofacial growth is a biological process which is regulated by intrinsic cellular factors and local factors. It is not actually clear yet how these elements influence each other.
An important concept to understand the dynamics of craniofacial growth, is the difference between remodeling and dislocation.
Dislocation physically creates the space needed for bone growth and this is possible thanks to the presence of sutures.
Remodeling, thanks to the selective activation of osteogenetic tissues, allows bony structures to achieve a certain shape and to keep it throughout the various phases of growth.
Remodeling processes allow adaptation of the achieved shape to the functional needs of the structures.
During growth, while bony segments are displaced, they also continually get remodeled (Enlow, 1975).
In CFS the equilibrium between remodeling and dislocation processes is severely altered.
SHORT REVIEW OF NORMAL GROWTH OF THE CRANIAL BASE AND OF THE CRANIAL VAULT
The development of the cranial base has a fundamental influence on the structure, the dimensions and the growth of the facial skeleton (Enlow, 1975).
The anterior segment of the cranial base (from basion to nasion) at birth is about 56% of its final dimension and at two years of age it reaches 70%.
The width of the cranial base in a newborn is about 100 mm, within six months it grows of another 50 mm and of another 20 mm within the first year of life.
Between the 3rd and the 14th year of life it will grow half a millimeter every 12 months.
At birth the inter-sphenoidal syncondrosis have almost ceased to grow, while the spheno-ethmoidal and the spheno-occipital syncondrosis remain active until about seven years of age (Fig. 10.1).
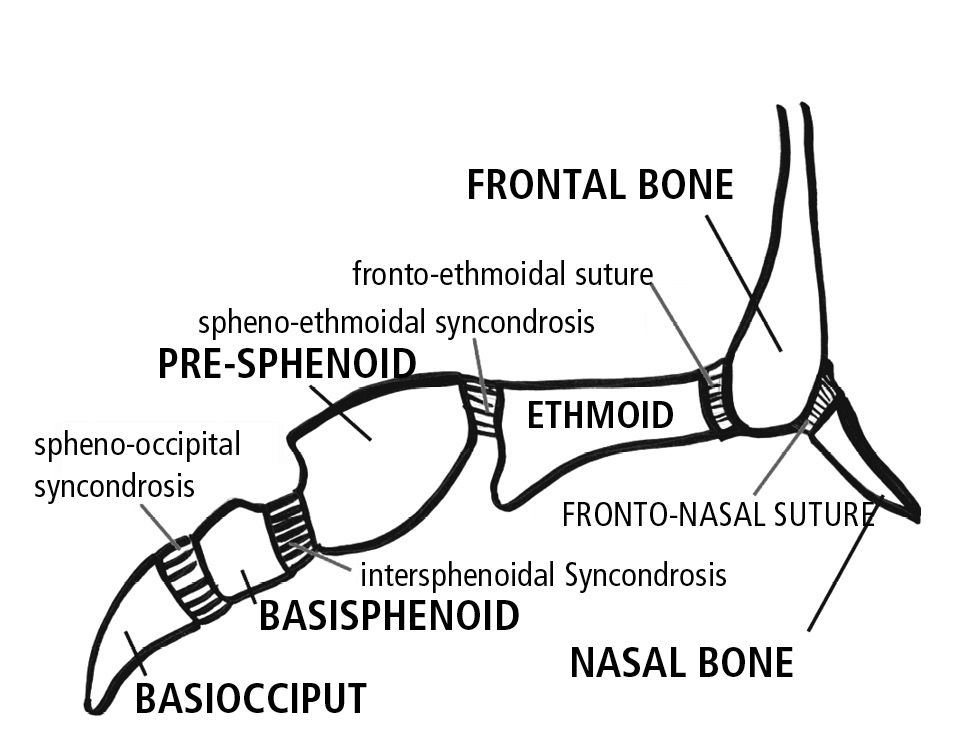
Fig. 10.1 • Syncondroses of the cranial base.
Cranial base growth has an intrinsic component, which is provided by the growth potential within the syncondrosis, but there is also an extrinsic component.
The extrinsic component, which corresponds to the functional matrix, derives from the interaction between the intrinsic sutural growth at their growth sites, and the thrust of the brain, which is expressed at the sutural and at the periosteal level.
The growth of the cerebral hemispheres, especially the frontal lobes and the temporal lobes, expands the medial and anterior cranial fossa.
The anterior fossa and the naso maxillary complex are pushed anteriorly by the frontal lobe, which moves forward thanks to the increasing size of the temporal lobe behind it.
The anterior cranial base presents resorption on its superior aspect and apposition on its inferior aspect with the results of an inferior repositioning of the anterior floor.
This happens also to the occipital base which has an intra cranial resorption and extra-cranial apposition.
In the cranial vault the exocranial surface is an area of bony apposition and the intra cranial surface is an area of resorption which allows for an increment in the volume of the cranium.
This equilibrium between apposition and resorption at the level of the cranial base is able to maintain the correct space for the nervous and vascular structures without dislocating them.
SHORT REVIEW OF NORMAL FACIAL GROWTH
There are two fundamental factors regulating the development of the craniofacial skeleton: the morphology of the cranial base and the anatomical and functional components of the craniofacial skeleton.
The morphology of the cranial base is a foundation for the facial skeleton, whereby it influences its proportions.
The expansion of the medial fossa induces an anterior movement of the anterior fossa and of the naso-maxillary complex.
The floor of the anterior cranial base is the direct counterpart of the maxillary bone, just as the dimension of the pharynx is guided by the dimension of the medial cranial fossa (Enlow, 1975).
The second factor, is directly linked to breathing, speech and feeding systems and to their active role during fetal growth and postnatal growth.
For example, in the neonate, the cranial vault, the cranial base and the orbital region are relatively large compared to the face and they are accompanied by rapid growth of the brain and of the orbital structures.
The facial skeleton, on the other hand, is small in the fetus, and increases its dimensions in postnatal life when breathing, suction, swallowing and dentition gradually become more relevant.
Ossification occurs, depending on the area, both with an intra-membranous mechanism and with an endochondral mechanism. Intra-membranous ossification is allowed by the presence of periosteum and sutures, while endochondral ossification occurs in those areas where a vascularized membrane, such as the periosteum, would not able to withstand the excessive pressure and therefore cartilage is used as a scaffold.
Syncondrosis allow growth that adapts to pressure, while sutures allow growth where there is tension.
As in the cranium, in the face there are areas of apposition and areas of resorption: at the orbital level, the maxillary, the palatal and the nasal level.
The major objective of facial growth is to allow for expansion of the upper airspace: the sinuses, the oral cavity, the pharynx and the choanae.
All these processes together guide an inferior and anterior growth of the maxilla with resorption on the anterior surface and bony apposition on the posterior surface (Fig. 10.2).
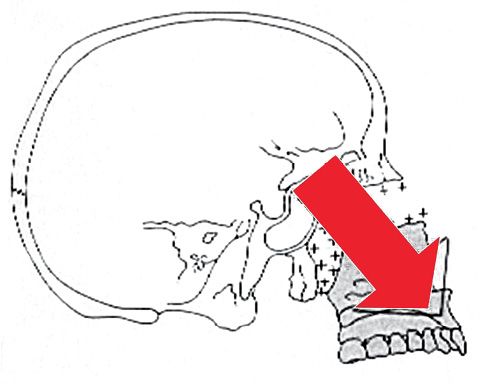
Fig. 10.2 • Forward and downward growth of the maxilla.
The nasal cavities present almost exclusively resorption surfaces which induce an expansion in a lateral direction and an inferior direction and an inferior movement of the palate (Fig. 10.3).
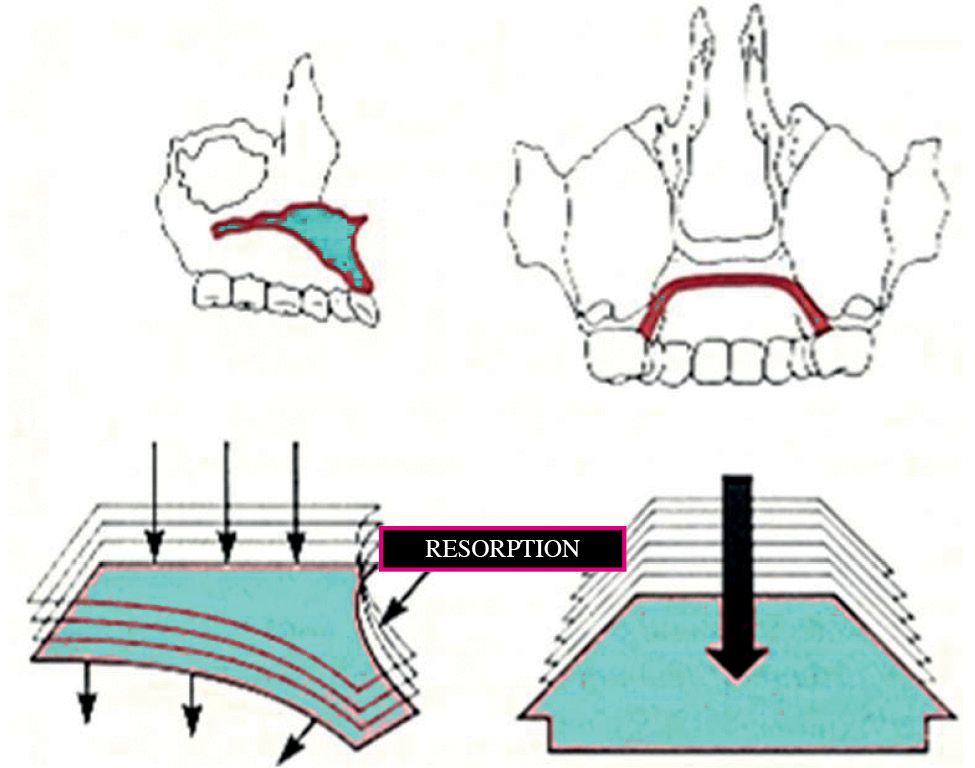
Fig. 10.3 • Vertical growth of the maxilla, with resorption on the nasall floor and apposition on the palatal aspect.
PRE AND POSTSURGICAL GROWTH IN CRANIOFACIAL SYNOSTOSIS (GROWTH POST LE FORT III OSTEOTOMIES)
In a study conducted on over 30 patients subjected to classical Le Fort III osteotomy (Meazzini, 2005), we have studied the growth of CFS patients not only after, but also prior to surgery.
The results have shown that there is no horizontal growth, but only vertical growth of the midface relative to anterior cranial base. This pattern continues unaltered after a Le Fort III osteotomy, suggesting that a Le Fort III has no influence on the pathological growth typical of CFS (Figg. 10.4a-d).
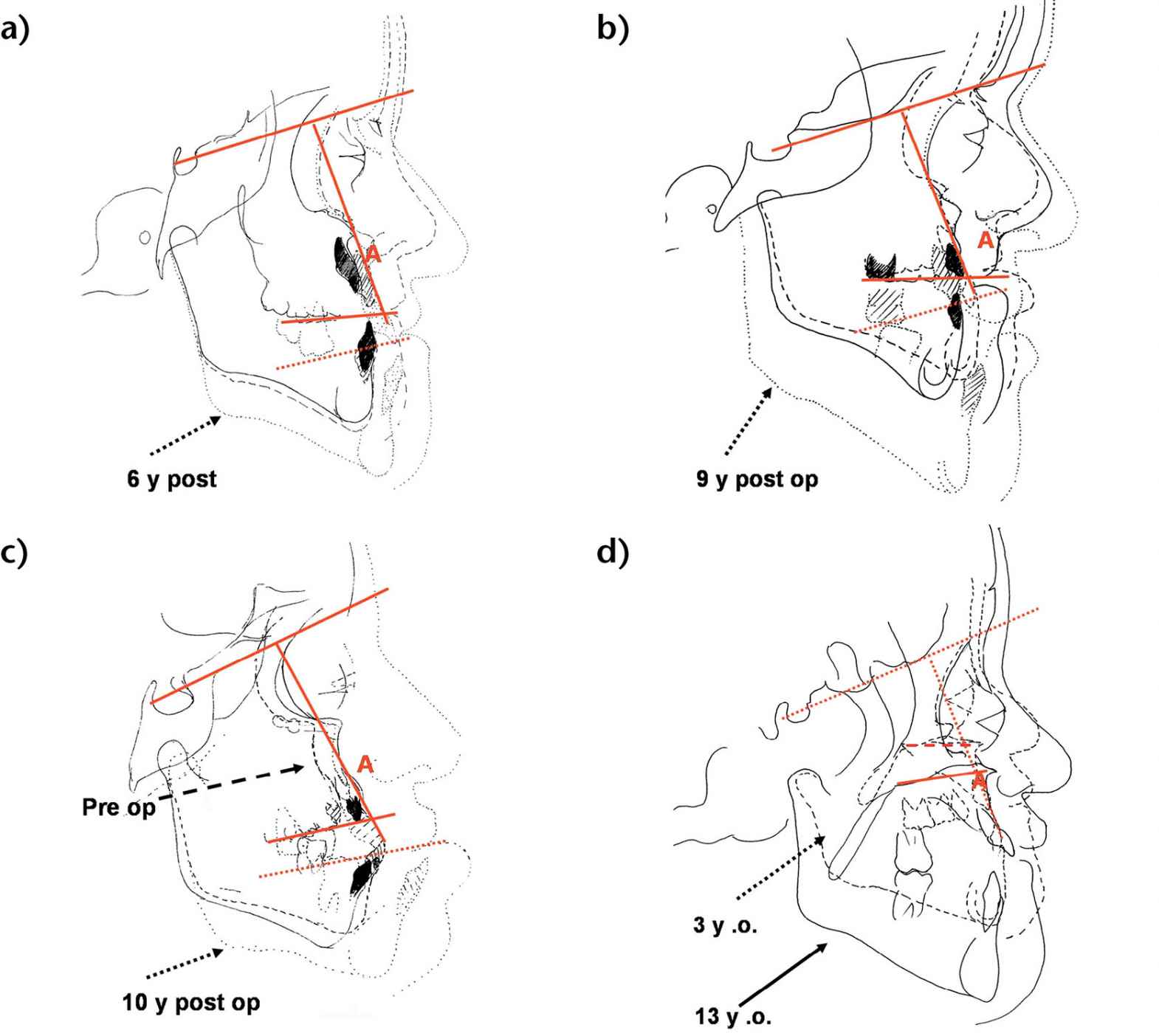
Figg. 10.4a-d • Cephalometric superimpositions of tracings of the patients pre-Le Fort III osteotomy and long term post le Fort III osteotomy (a. 6 years post surgery, b. 9 years; c. 10 years). d. Cephalometric superimpositions of tracings of an Apert patient at 3 years and at 13 years never operated. Note the absence of horizontal growth and presence of vertical growth relative to anterior cranial base, both in the non operated patient and in the post Le Fort III growth.
Tessier (1971) had initially hoped that after a Le Fort III, maxillary growth would be “unleashed”. This has been shown not to be true, though some controversy still stands on whether this type of surgery inhibits subsequent growth (Bachmeyer, 1980; Kaban, 1986).
In particular, some authors suggest that scar tissue (Shapiro, 1981) or periosteal damage (Squier, 1982) may inhibit skeletal growth. While some authors have suggested that after Le Fort III osteotomy the growth of CFS patents is hindered, if compared to non-operated CFS patients (Bachmeyer, 1986), other authors have suggested that the growth potential of CFS patients is not influenced by a Le Fort III osteotomy (Kreiborg, 1986; David, 1990; Meazzini, 2005).
CLINICAL FEATURES
APERT SYNDROME
In 1557 Licostene described a child with a cranial deformity and a deformity of hands and feet, it was an acrocephalosyndactily, later described by Apert in 1906.
Apert syndrome is a congenital syndrome with autosomic dominant transmission and it is characterized by craniosynostosis and a severe syndactily of hands and feet (Fig. 10.5) (Upton, 1991; Ibrahimi, 2001).
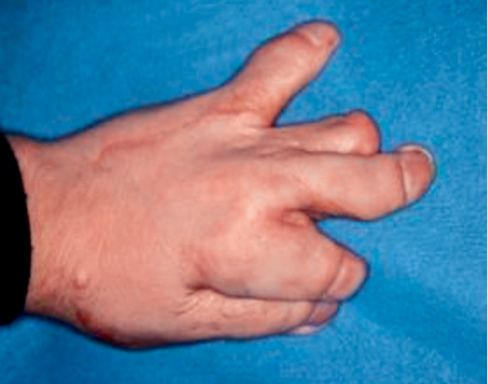
Fig. 10.5 • Syndactily in a child affected by Apert Syndrome.
Seldom this syndrome is associated to anomalies of the central nervous system, of the cardiovascular system and of the skin (Oldrige, 1999).
The incidence of Apert syndrome is around 12.4 patients over 1 million of new births and it is more frequent in Asia (22.3 cases per million); males and females are equally affected (Tolarova, 1997).
In the majority of patients the syndrome is sporadic, possibly linked to a mutation of a paternal allele. These mutations might be correlated with the age of the father, which might be considered a risk factor (Cohen, 1988; Tolarova, 1997).
Apert syndrome is also called Acrocephalosyndactily, as the craniofacial synostosis is characterized by acrobrachycephaly, midfacial hypoplasia and symmetrical syndactily, both bony and cutaneous.
The syndactily generally involves the second, the third and the fourth finger (Cohen, 1975 and 1992).
The most frequent synostosis in Apert syndrome is that of the coronal suture bilaterally. Usually the spheno-zygomatic, the spheno-temporal and the occipital-mastoid sutures are still radiologically evident at birth.
The zygomatic processes of the frontal bone are hypoplastic, the cranial base is deformed and often asymmetrical, the anterior cranial fossa is very short with shallow orbits, which, together with the hypoplasia of the midface, are the cause of a severe exorbitism.
There is an increased bitemporal diameter as a consequence of the compensatory growth of the squamous sutures.
The cranium is short sagittally, the forehead is tall and the occiput is flat.
As the extension and the timing of the synostosis is variable, the cranial aspect may be different in different patients, we may see turricephaly, brachycephaly or plagiocephaly.
The increment in width of the frontal sinuses and the ethmoidal sinuses determines the hypertelorism (Figg. 10.6a-b).
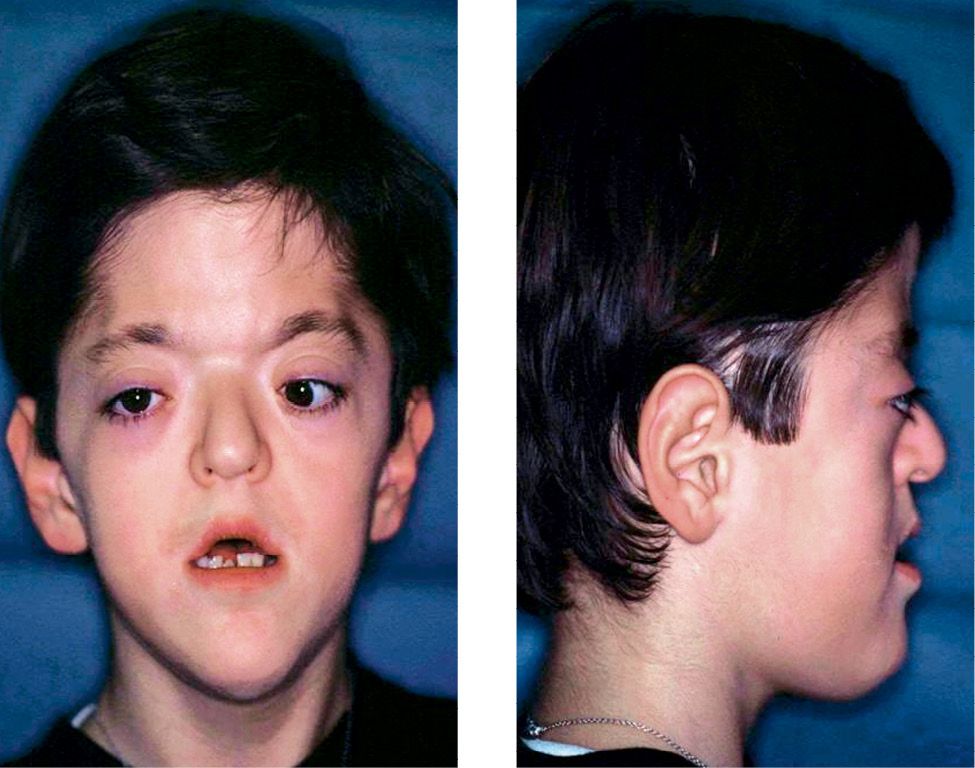
Figg. 10.6a-b • Facial features in a child affected by Apert Syndrome. Acrobrachycephaly, midfacial hypoplasia, exorbitism, hypertelorism, parrot beak nose.
Generally the naso-frontal angle is acute, the nose has a parrot beak shape, the nasal septum is often deviated and the palatal vault is high and narrow. The hard palate is shorter, but the soft palate is longer than normal and in 30% of cases there is a soft palatal cleft.
The maxillary arch is crowded and it has a characteristic “v” shape and its vault is thickened by deposits of glycosaminoglycans (Fig. 10.7).
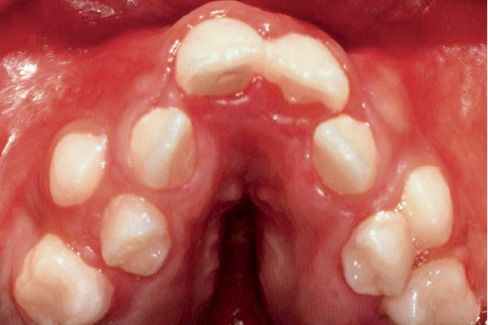
Fig. 10.7 • Severe crowding, V shaped arch and palatal vault thickened by deposits of glycosaminoglycans in a child affected by Apert Syndrome.
A choanal atresia or hypoplasia may be associated, incrementing the risk of respiratory distress and sleep apnoeas (Lowe, 2000).
The abnormalities at the maxillary level cause a class III malocclusion, with anterior open bite and bilateral posterior cross bite (Fig. 10.8).
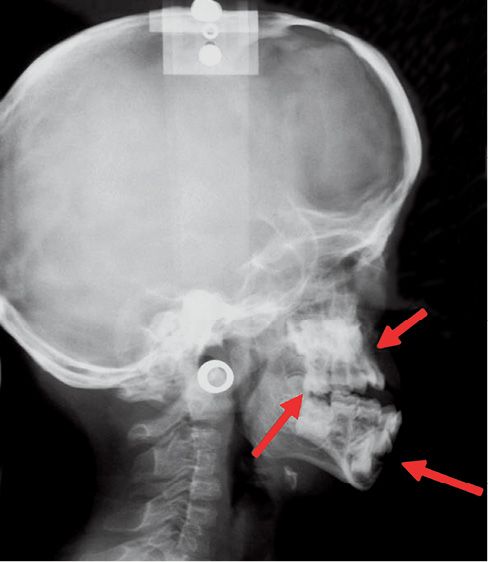
Fig. 10.8 • Skeletal characteristics of a patient affected by Apert Syndrome. Midfacial retrusion, open bite and pseudo-prognathism of the mandible.
Regarding the cranial base, the floor of the anterior cranial fossa has an excessive inclination and there is a deformity in the cranial base flexure which is correlated to the exorbitism and to the midface hypoplasia (Rosen, 1984).
In the majority of the patients with Apert syndrome no mental retardation is reported (Olridge, 1999; Kai Yu, 2000).
From the genetic standpoint Apert syndrome presents mutations of genes that control fibroblast growth factors receptors, in particular FGFR2.
The effect of these mutations are essentially of two types: on one hand the growth factors are less able to dissociate from the receptors (Anderson,1998), on the other hand the receptors lose their specificity for their ligands. This has the effect of a hyper activation of the FGFR2 receptor (Kai Yu, 2000).
The consequences of this hyper activation are evident on the intramembranous ossification of the cranium (Wilkie, 1987).
Immature osteoblasts are stimulated by fibroblast growth factors to proliferate and accelerate ossification as well as accelerate the process of completion of ossification of the sutures themselves.
CROUZON SYNDROME
Crouzon syndrome is a disease with an autosomic dominant transmission, also associated to mutations of the gene that codes for FGFR2. Clinically it is characterized by craniosynostosis, midfacial hypoplasia, shallow orbits and exorbitism (Lowe, 2000) (Fig. 10.9a-b).
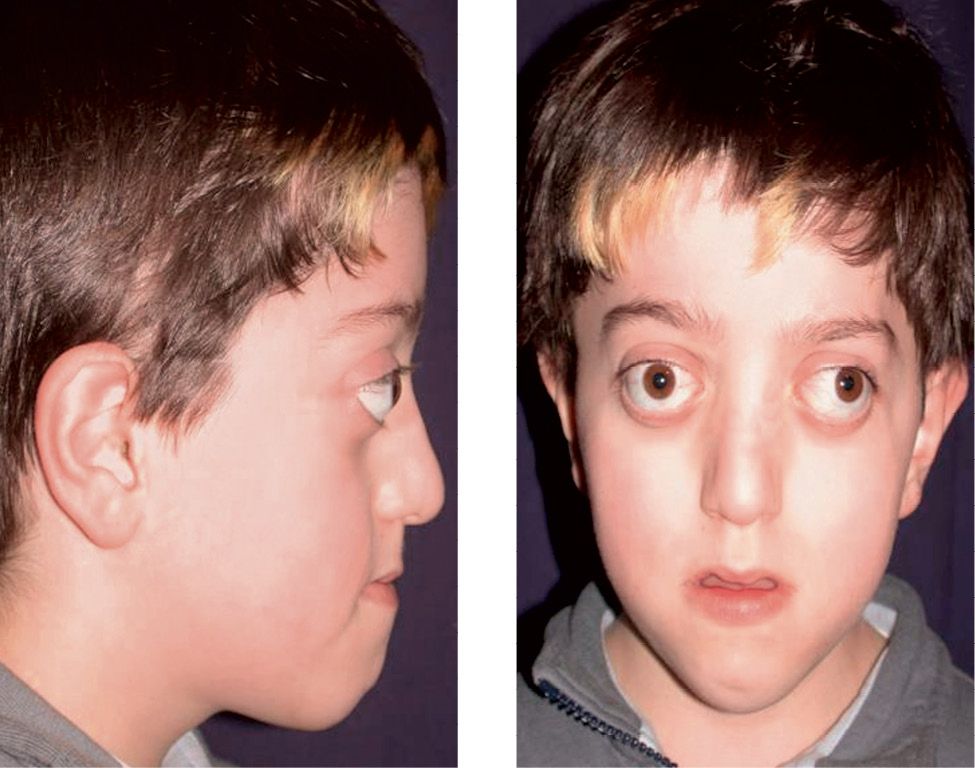
Figg. 10.9a-b • Facial features in a child affected by Crouzon Syndrome. Brachycephaly, midfacial hypoplasia, severe exorbitism, hypertelorism.
Its incidence is of 16.5 new borns over 1 million; it represents about 4.8% of the diagnosed craniofacialsynostosis (Tsai, 2003). It was first described by Crouzon in 1912, as a syndrome where the synostosis of the coronal sutures or the sagittal suture had a consequent sagittal hypo-development of the cranial base, especially the anterior cranial base, and a secondary severe sagittal and vertical development of the midface. The phenotype is therefore an acrobrachycephaly with a severe protrusion of the midface, exorbitism, related especially to the midfacial hypoplasia and of the roof of the orbits. The occlusion is a class III malocclusion with peudo prognathism. The open bite is not as severe as in Apert syndromes, and the crowding, though severe, is usually less challenging than in Apert syndrome. The palate is narrow, and seldom atresia of the coanae or stenosis of the hearing canals may be present. There is an increased inclination of the anterior cranial fossa and of the cranial base flexure which are correlated with the exorbitism and the midfacial hypoplasia (Rosen, 1984). Cervical vertebrae fusions may be present, and elbows deformities may be associated (Lowe, 2000).
CROUZONODERMOSKELETAL SYNDROME
Crouzonodermoskeletal syndrome is an autosomal dominant disorder characterized by craniosynostosis and acanthosis nigricans. Mutations in the FGFR3 gene cause this syndrome, which is very similar to Crouzon. Signs in common with Crouzon syndrome are exorbitism, hypertelorism, strabismus, midface hypoplasia, beaked nose. In addition, Crouzonodermoskeletal syndrome patients are affected by acanthosis nigricans, a skin condition characterized by thick, dark skin in all body folds. Mandibular cementomas may develop during young adulthood.
JACKSON-WEISS SYNDROME
Jackson-Weiss syndrome is a rare genetic autosomal dominant disorder characterized by craniosynostosis associated to feet abnormalities caused by mutations in the FGFR2 gene. Its incidence is unknown. As in Crouzon syndrome hypertelorism and exorbitism are a common facial feature. Foot abnormalities, with short and wide toes, are the most consistent features. Syndactily may be present. The hands are almost always normal.
CARPENTER SYNDROME
Carpenter syndrome, also called acrocephalopolysyndactyly type II, is an extremely rare autosomal recessive congenital disorder associated with mutations in the RAB23 gene. Its incidence is 1 in 1 million live births. The syndrome is characterized by scaphocephaly or brachycephaly, exorbitism, cutaneus syndactily or polydactily of hands and feet. Mental delay is also frequent (75%). Obesity is a distinctive symptom. Facial features include flat nasal bridge, wide upturned nose with abnormally large nasal nostrils, low set deformed ears. Maxillary retrusion with a high arched and narrow palate with delayed tooth eruption and peg shaped teeth. Heart anomalies are present in 50% of the patients.
PFEIFFER SYNDROME
PFEIFFER SYNDROME
This is an autosomic dominant syndrome with variable expression, associated to mutations of FGFR2 and 1 (Cohen, 1993). Pfeiffer syndrome affects about 1 in 100,000 individuals.
It was first described by Pfeiffer in 1964. The synostosis of the coronal suture causes a brachycephaly. Severe hypoplasia of the midface, hypertelorism are present. The patient with Pfeiffer’s syndrome has a similar craniofacial deformity to that of the Apert’s and Crouzon’s syndromes patients, however, the midface in Pfeiffer’s syndrome patients is usually more severely affected.
About 50% of children with Pfeiffer syndrome have hearing loss. Bracydactily, with broad thumbs and toes are present and occasionally, a partial syndactyly involving the second and third digits (Cohen, 1993). (Figg. 10.10 a-b and 10.11).
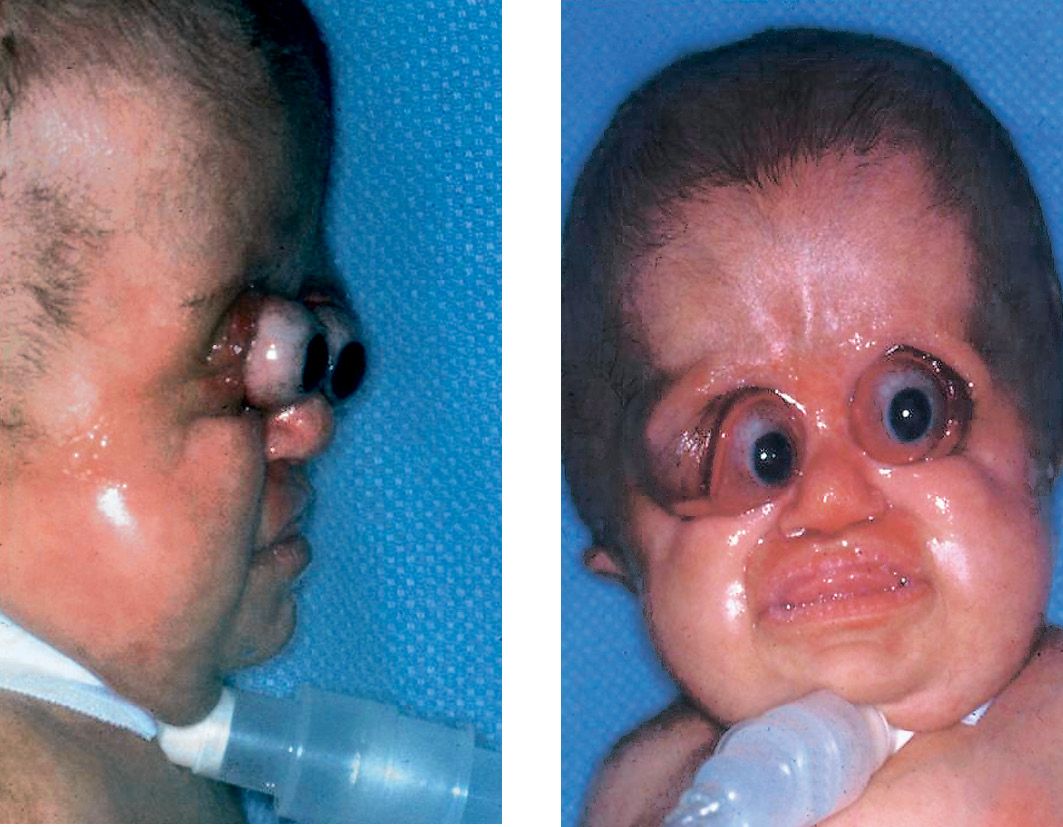
Figg. 10.10a-b • Facial features in a child affected by Pfeiffer Syndrome. Midfacial hypoplasia, severe exorbitism. Note in this patient the severe globe luxation and the presence of a severe respiratory distress requiring tracheostomy (Courtesy of Drs. Polley and Figueroa, Chicago).
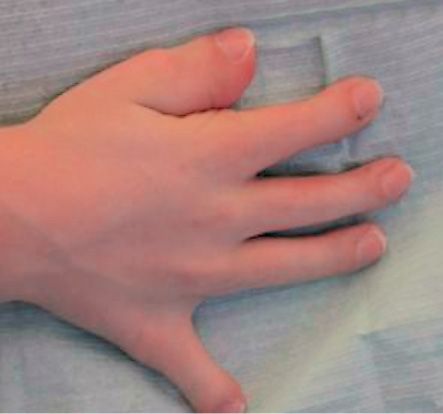
Fig. 10.11 • Brachydactily in a child affected by Pfeiffer Syndrome.
Depending on the severity of the symptoms this syndrome has been classified in three types (Schell, 1995; Robin, 1998).
Type 1 or “classic” Pfeiffer syndrome has symptoms as described above. Most individuals with type 1 have normal intelligence and a normal life span.
Types 2 and 3 are more severe forms of Pfeiffer syndrome, often involving problems with the nervous system. Type 2 is distinguished from type 3 by the more common incidence of pansynostosis, leading to a “cloverleaf” shaped head.
SAETHRE-CHOTZEN SYNDROME
Saethre-Chotzen syndrome is an autosomal dominant genetic condition caused by mutations in the TWIST1 gene. It is characterized by coronal synostosis. Facial features include brachycephaly, a low frontal hairline, eyelid ptosis, hypertelorism and a broad nasal bridge. Facial asymmetry is always present. It has an estimated prevalence of 1 in 25,000 to 50,000. Most patients with Saethre-Chotzen syndrome also present hipotia or ear deformity. The signs and symptoms of Saethre-Chotzen syndrome vary widely, even among affected individuals in the same family. This condition can also be associated with abnormalities of hands and feet, such as cutaneus syndactily between the second and third fingers on each hand and a broad or duplicated toes. Less common signs and symptoms of Saethre-Chotzen syndrome include short stature, vertebral abnormalities, hearing loss, and heart defects.
Robinow syndrome is a condition with features similar to those of Saethre-Chotzen syndrome, including craniosynostosis and broad or duplicated great toes. It was found to result from mutations in the same gene and is now thought to be a mild variant of Saethre-Chotzen syndrome.
REFERENCES – EMBRYOLOGY, GROWTH AND CLINICAL FEATURES
Anderson 1998 Anderson J, Burns HD, Enriquez-Harris P, Wilkie AO, Heath JK. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. School of Biochemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. Hum Mol Genet. 1998 Sep; 7(9): 1475-83.
Apert 1906 Apert ME. De l’acrocephalosyndactylie. Bull Mem Soc Med Hop Paris 1906; 23: 1310-1330.
Bachmayer 1986
1) Bachmayer DI, Ross RB. Stability of Le Fort III advancement surgery in children with Crouzon’s, Apert’s, and Pfeiffer’s syndromes. Cleft Palate J. 1986 Dec: 23 Suppl 1: 69-74.
2) Bachmayer DI, Ross RB, Munro IR. Maxillary growth following LeFort III advancement surgery in Crouzon, Apert, and Pfeiffer syndromes. Am J Orthod Dentofacial Orthop. 1986 Nov; 90(5): 420-30.
Cohen 1975 Cohen MM Jr. An Etiologic and nosologic overview of craniosynostosis syndromes. Birth Defects Orig Artic Ser 1975; 11(2): 137-89. Review.
Cohen 1988 Cohen MM. Craniosynostosis update 1987. Jr Am J Med Genet Suppl 1988; 4: 99-148. Review.
Cohen 1992
1) Cohen MM Jr, Kreiborg S. Upper and lower airway compromise in the Apert syndrome. Am J Med Genet. 1992 Sep 1; 44(1): 90-3.
2) Kreiborg S, Barr M Jr, Cohen MM Jr. Cervical spine in the Apert syndrome. Am J Med Genet. 1992 Jul 1; 43(4): 704-8.
3) Cohen MM Jr, Kreiborg S. New indirect method for estimating the birth prevalence of the Apert syndrome. Int J Oral Maxillofac Surg. 1992 Apr; 21(2): 107-9.
4) Cohen MM Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erickson JD, Roeper P, Martínez-Frías ML. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992 Mar 1; 42(5): 655-9.
5) Kreiborg S, Cohen MM Jr. The oral manifestations of Apert syndrome. J Craniofac Genet Dev Biol. 1992 Jan-Mar; 12(1): 41-8.
Cohen 1993 Cohen MM Jr. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet. 1993 Feb 1; 45(3): 300-7.
Cornejo-Roldan 1999 Cornejo-Roldan LR, Roessler E, Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Medical Genetics Branch, National Human Genome Research Institute, Bethesda, MD, USA. Hum Genet. 1999 May; 104(5): 425-31.
David 1990 David DJ Sheen R. Surgical correction of Crouzon Syndrome. Plast reconstr Surg 1990 Mar, 85(3): 344-54.
Enlow. Enlow D.H. Handbook of facial growth. 1975.
Hajihosseini 2001 Hajihosseini MK, Wilson S, De Moerlooze L, Dickson C. A splicing switch and gain-of-function mutation in FgfR2-IIIc hemizygotes causes Apert/Pfeiffer-syndrome-like phenotypes. Proc Natl Acad Sci U S A. 2001 Mar 27; 98(7): 3855-60.
Ibrahimi 2001 Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci U S A. 2001 Jun 19; 98(13): 7182-7. Epub 2001 Jun 5.
Kaban 1986 Kaban LB, Conover M, Mulliken JB. Midface Position after Le Fort III advancement: a long-term follow-up study. Cleft Palate J, 1986 Dec; 23 suppl 1: 75-7.
Kai Yu 2000 Yu K, Herr AB, Waksman G, Ormitz DM. Loss of fibroblast growth factor receptor 2 ligand binding specificity in Apert syndrome. Proc Natil Acad Sci USA 2000 Dec 19; 97(26): 14536-41
Kreiborg 1986 Kreiborg S, Aduss H. Pre- and postsurgical facial growth in patients with Crouzon’s and Apert’s syndromes. Cleft Palate J. 1986 Dec; 23 Suppl 1: 78-90.
Lowe 2000 Lowe LH, Booth TN, Joglar JM, Rollins NK. Midface anomalies in children. Radiographics. 2000 Jul-Aug; 20(4): 907-22; quiz 1106-7, 1112. Erratum in: Radiographics 2000 Sep-Oct; 20(5): 1494.
McCarthy 1990 McCarthy, La Trenta GS, Breitbart AS, Grayson BM, Brookstein FL. The Le Fort III advancement osteotomy in the child under 7 year of age. Plast Reconstr Surg. 1990 Oct 86(4) 633-46 discussion 647-9.
Meazzini 2005
1) Meazzini MC, Mazzoleni F, Gabriele C, Bozzetti A. Mandibular distraction osteogenesis in hemifacial microsomia: long-term follow-up. J craniomaxillofac Surg. 2005 Dec, 33(6): 370-6.
2) Meazzini MC, Mazzoleni F, Caronni E, Bozzetti A. Le Fort III advancement osteotomy in the growing child affected by Crouzon’s and Apert’s syndromes: presurgical and postaurgical qrowth.J Craniofac Surg 2005. May, 16(3): 369-77.
Muenke 1998 Gripp KW, Stolle CA, McDonald-McGinn DM, Markowitz RI, Bartlett SP, Katowitz JA, Muenke M, Zackai EH. Phenotype of the fibroblast growth factor receptor 2 Ser351Cys mutation: Pfeiffer syndrome type III. Am J Med Genet. 1998 Jul 24; 78(4): 356-60.
Oldridge 1999 Oldridge M, Zackai EH, McDonald-McGinn DM, Iseki S, Morriss-Kay GM, Twigg SR, Johnson D, Wall SA, Jiang W, Theda C, Jabs EW, Wilkie AO. De novo alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am J Hum Genet. 1999 Feb; 64(2): 446-61.
Robin 1998 Robin NH, Scott JA, Arnold JE, Goldstein JA, Shilling BB, Marion RW, Cohen MM Jr. Favorable prognosis for children with Pfeiffer syndrome types 2 and 3: implications for classification.Center for Human Genetics, Department of Genetics, Case Western Reserve University School of Medicine, Cleveland, Ohio 44106, USA. Am J Med Genet. 1998 Jan 23; 75(3): 240-4.
Roche 1977 Roche AF, Lewis AB, Weiner H, Mc Cartin R. J. Late elongation of the cranial base. Dent Res 1977 Jul; 56(7): 802-8.
Rosen 1984 Rosen HM, Whitaker LA. Cranial base dynamics in craniofacial dysostosis. J Maxillofac Surg. 1984 Apr; 12(2): 56-61.
Schell 1995 Schell U, Hehr A, Feldman GJ, Robin NH, Zackai EH, de Die-Smulders C, Viskochil DH, Stewart JM, Wolff G, Ohashi H, e col. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum Mol Genet. 1995 Mar; 4(3): 323-8.
Shapiro 1981 Shapiro PA, Kokich VG, Hohl TH, Loe C. The effect of early Le Fort I osteotomies on craniofacial growth of juvenile Macaca nemestrina monkeys. Am J Orthod 1981 May, 79(5): 492-9.
Squier 1982 Squier CA, Kremenak CR. Quantitation of the healing palatal mucoperiosteal wound in the beagle dog. Br J Exp Pathol 1982 Oct, 63(5): 573-84.
Tessier 1971
1) Tessier P. Total osteotomy of the middle third of the face for faciostenosis or the sequelae of Le Fort 3 fractures. Plast Reconstr Surg. 1971 Dec, 48(6): 533-41.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


