Hal S. Meltzer, MD and Michael L. Levy, MD, PhD
13.1 Introduction
Craniosynostosis can be defined as the premature fusion of one or more sutures of the skull.1 When skull sutures fuse prematurely as an isolated event, the process is referred to as nonsyndromic craniosynostosis.2 When skull sutures fuse in conjunction with other anomalies in a clinically recognizable pattern, the process is referred to as syndromic craniosynostosis.1 There have been as many as 90 reported syndromes that are associated with craniosynostosis. The most common of these syndromes are Apert’s, Crouzon’s, Pfeiffer’s, Saethre–Chotzen, and Carpenter’s.1 These syndromes are usually genetic and can be inherited via autosomal dominant, autosomal recessive, or X-linked patterns. Although there are some key distinguishing features for each syndrome, there are many more common traits. These include multiple-suture craniosynostosis, midface retrusion, exorbitism, and limb anomalies.1
The etiology of craniosynostosis remains unclear. However, molecular genetic analysis has helped to identify a relationship between the above syndromes and an abnormal gene encoding for fibroblast growth factor.1,2 There is still debate about the exact relationship between the cranial base synchondroses and the cranial vault sutures. Some argue that the primary issue lies within the cranial base, thus resulting in abnormal fusion in the cranial vault. Others believe that the cranial vault is at fault, which translates into abnormal cranial base and subsequent abnormal facial development.1–3
13.2 Functional Issues in Craniosynostosis
Normal craniofacial growth depends on patent sutures between the cranial vault and base bones as well as the presence of a rapidly expanding brain.1,2 The brain typically triples in size during the first year of life and continues to grow at a high rate until approximately 6 years of age. As the brain grows, it displaces the overlying cranial vault bones which, in turn, grow and remodel. If the cranial vault sutures are fused during this rapid growth phase of the brain, growth and development are unable to occur, and functional issues may develop.1,2 One of the more significant of these issues is an increased intracranial pressure (ICP). This problem develops as a result of a discrepancy between brain size and cranial vault volume. Clinical symptoms suggesting raised ICP include headaches, irritability, and difficulty sleeping. Clinical signs include papilledema on funduscopy, and luckenschadel (beaten metal) appearance of the inner table of the vault as seen in plain radiography and computed tomography (CT).1,2,4 This abnormal appearance of the inner table of the vault is, however, a late finding. Marchac et al. report that up to 42% of children with multiple-suture craniosynostosis will have intracranial hypertension. This is markedly higher than their reported incidence of 13% in those children with single-suture synostosis.5 The diagnosis of raised ICP is often made clinically, since the signs on CT are often unreliable.1,2
Disturbance of vision can accompany multiple-suture synostosis and can manifest by three main mechanisms. The first is a result of exorbitism, which is defined as the displacement of the globe from its normal position due to underdeveloped or abnormally shaped orbits. This is a result of the severe midface and orbital hypoplasia that often occurs within the syndromes. This exorbitism can result in corneal exposure and keratitis, which can in turn lead to infection, pain, ulceration, or even blindness. The second mechanism of visual disturbance is a result of ocular dysmotility secondary to the abnormal shape and size of the orbits and extraocular muscles. This form of disturbance will often present as strabismus or exotropia. The third mechanism relates to the aforementioned raised ICP. Intracranial hypertension can lead to optic atrophy and blindness as a result of diminished vascular perfusion to the retina.1,2,6
Another issue that can arise in this patient population is hydrocephalus. The most popular theory explaining it is, that increased venous pressure within the sagittal sinus as a result of craniosynostosis prevents outflow from the sinus. The diminished outflow can result in communicating hydrocephalus or, less commonly, the noncommunicating form. Hydrocephalus can be identified either clinically, with findings similar to those of raised ICP, or radiographically, as enlarged ventricles.4
The final functional issue that can develop is neuropsychiatric disturbance.7 This can present on a spectrum, from mild behavioral abnormality to overt mental retardation. Several studies have demonstrated differing results when it comes to mental retardation associated with craniosynostosis and the role of surgery in preventing this issue. It is generally accepted that the rate of mental dysfunction in children with both syndromic and nonsyndromic craniosynostosis is higher than in the normal population. However, it remains disputed whether the primary cause of this issue is a result of a poorly developed brain or of a calvaria restricting the growth of an otherwise normal brain.4,7 The former is thought to be more likely in the setting of a single-suture craniosynostosis.4 It is also important to consider the psychological disturbance that can be involved with having a severely misshapen calvaria.7
13.3 Nonsyndromic Craniosynostosis
The major sutures of the cranial vault include the metopic, sagittal, coronal, and lambdoid.2 “Nonsyndromic craniosynostosis” encompasses fusion of one of these sutures in a sporadic occurrence without any other systemic abnormalities. Assessment of the patient with an abnormal head shape should focus on obtaining a focused history, performing an appropriate physical examination, and acquiring any necessary diagnostic imaging studies. Finally, it should involve consultation with a geneticist, neurosurgeon, and/or ophthalmologist.2
Important points to elicit on history include family history of craniosynostosis, as well as signs and symptoms of raised ICP (nausea, headache, irritability). Genetic testing and counseling can help to both establish a diagnosis and inform the parents of current and future issues. Physical examination should involve palpating the cranial vault sutures for ridging and the fontanelles for patency and bulging. Close attention should be paid to the facial and calvarial morphology. The eye and periorbital structures should be evaluated. Jaw size and position, including occlusion, should be noted. Acquiring diagnostic images, including 3-dimensional CTs, continues to be controversial for patients with isolated, single-suture craniosynostosis.2 It is common practice at our center to obtain a CT with 3-dimensional reconstructions because we feel it is important not only for assessing the underlying brain, but also for aiding in perioperative reconstruction (Figure 13.1).
The plagiocephaly associated with craniosynostosis must be distinguished from the plagiocephaly associated with external, positional factors (positional plagiocephaly).2,7 That deformity is the result of external pressure on the developing skull, either in utero or in early infancy, leading to an abnormal head shape that often mimics craniosynostosis. This distinction is important because, although operative intervention is the standard of care for most moderate to severe craniosynostotic deformities, nonoperative helmet-molding therapy is typically used for most positional deformities. The distinction between these two entities can usually be made during the physical examination. Signs of positional plagiocephaly include absent cranial suture ridging, patent fontanelles, and the parallelogram head deformity, which is characterized by ipsilateral occipital flattening and frontal bossing with an anteriorly displaced ear.2,7
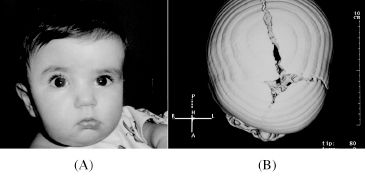
Figure 13.1. (A) Photograph of a child with right unicoronal craniosynostosis. (B) 3-dimensional CT image of the same child. Notice the patent anterior fontanelle and left unicoronal suture, along with the fused right unicoronal suture.
13.3.1 Types of synostosis
Listed below are the major types of cranial vault synostoses and their associated characteristics.
13.3.1.1 Sagittal synostosis
Affected suture: Sagittal
Resulting deformity: Scaphocephaly
Incidence: 1 in 2,000 (40% of all nonsyndromic craniosynostosis) Epidemiology: Sporadic, male:female of 4:1
Physical examination: Palpable midline ridge with narrowing of the skull in a biparietal and bitemporal dimension. Frontal and occipital bossing are also present.2,7
13.3.1.2 Metopic synostosis
Affected suture: Metopic
Resulting deformity: Trigonocephaly
Incidence: 1 in 2,500 to 15,000 (10–20%; recent studies have shown an increase of metopic synostosis up to 40% of all nonsyndromic craniosynostosis)
Epidemiology: Sporadic, male:female of 3:1
Physical examination: Can present on a spectrum of deformities ranging from a palpable mid-forehead ridge to a severely pointed forehead associated with bitemporal narrowing, biparietal flaring, and hypotelorism.2,7
13.3.1.3 Unicoronal synostosis
Affected suture: Coronal
Resulting deformity: Anterior plagiocephaly
Incidence: 1 in 10,000 (20% of all nonsyndromic craniosynostosis) Epidemiology: Sporadic, female:male of 3:2
Physical examination: Ipsilaterally, there is widening of the palpebral fissure, elevation and anterior displacement of the ear, nasal root deviation to the affected side, a prominent malar eminence, and an elevated and recessed supraorbital bar. Contralaterally, there is frontal bossing and the chin is usually deviated to the unaffected side.
Imaging: The harlequin orbit is present due to a lack of descent of the greater wing of the sphenoid. This deformity consists of loss of lateral orbital height with an elevated and recessed supraorbital bar.2,7
13.3.1.4 Bicoronal synostosis
Affected suture: Coronal
Resulting deformity: Turribrachycephaly
Incidence: 1 in 10,000 (20% of all nonsyndromic craniosynostosis) Epidemiology: Sporadic, male:female of 1:1
Physical examination: The skull is typically shortened in an anterior/ posterior dimension. There tends to be a widened forehead with hypoplastic supraorbital rims.1,2,7
13.3.1.5 Lambdoid synostosis
Affected suture: Lambdoid
Resulting deformity: Posterior plagiocephaly
Incidence: Rare, less than 3% of all nonsyndromic craniosynostosis Epidemiology: Sporadic, male:female of 2:1
Physical examination: Ipsilaterally, there is occipital flattening and an occipitomastoid boss, and the ear is displaced inferiorly and posteriorly. Contralaterally, there is a parietal and occipital boss as well as an occipitofrontal boss.2,7
13.3.2 Surgical treatment strategies
Many variables go into the decision-making process regarding the type of surgical intervention for nonsyndromic craniosynostosis. They include the age of the patient, the location and degree of the deformity, the overall health of the patient, and the potential need for repeat surgery.7–9 Generally speaking, limited (endoscopic, linear craniectomy) approaches are typically reserved for properly selected younger patients (less than 6 months old) with mild deformities affecting only one suture.10 These procedures rely on the growth of the underlying brain to aid in vault reshaping and often employ postoperative helmet-molding therapy to facilitate appropriate changes.10 The larger, more invasive approaches (open cranial vault remodeling with fronto-orbital advancement) are usually used for older children (older than 6 months) who have more severe deformities affecting one or more sutures.11 The open procedures often involve removal of the affected bones, with osteotomies and adjustments of those bones performed on a side table in the operating room. These remodeled bones are then placed back onto the exposed dura in a more anatomic position and stabilized in place using a number of different methods (suture, wire, plates, screws).11 The modern trend of fixation involves resorbable plating systems that are beneficial in that they (1) do not need to be removed since they are resorbed over time and (2) are less likely than titanium plates to interfere with facial growth. The main features of any operation for craniosynostosis include (1) releasing of the fused sutures, and (2) remodeling the hypoplastic and compensatory abnormalities of the craniofacial skeleton.11
It is generally agreed that in cases where functional issues are present (raised ICP, globe exposure), early intervention is the standard of care.5,8,12–15 In the absence of these functional issues, advocates of early surgery believe that a better, more naturally shaped skull can be obtained by reshaping the vault during early infancy, when the brain is in its most rapid-growth phase.8,15 Advocates of delayed surgery believe that the risk of blood loss and other complications is greater in early infancy, and that the deformity is more likely to recur if dealt with too early.6,14 Another consideration regarding the timing of operative intervention has to do with bone formation and development. Early in infancy, bone is very malleable and easier to remodel than it is beyond 1 year of age. The surrounding soft tissue of the infant cranium is also very osteogenic and permits the surgeon the flexibility of not having to fill in all bony gaps. These factors can lead to a technically easier surgery and perhaps a better result.12–14
The most important preoperative consideration for most skull-altering procedures involves dealing with blood loss. All patients must have blood products prepared preoperatively in case of significant loss during surgery.16–18 Optimization of medical comorbid conditions is critical, with appropriate consultations made well before the planned surgery.1,2
Choice of patient positioning intraoperatively involves providing the most complete access to the involved portions of the skull. The most common incision is the bicoronal incision. It is typically created in a zigzag fashion to help hide its appearance within the hair, especially in the temporal regions.19,20
Air embolism is a concern whenever the head is in a position above the heart. Cardiac Doppler monitoring as well as end-tidal carbon dioxide measurement are useful tools for detecting air entering the blood stream during large craniofacial operations.2,20
There are several different approaches to the various deformities in craniosynostosis. Outlined below is our center’s approach to each form of nonsyndromic craniosynostosis.
13.3.2.1 Sagittal synostosis
Endoscopic linear craniectomy of the fused sagittal suture is performed in those children presenting early (less than 3 months of age) with a mild to moderate deformity.11,21,22 Our goal is to release the fused suture at around 3 months of age, followed by placement of the child into helmet-molding therapy by 1 week postoperatively (Figures 13.2–13.8).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


