Introduction
The objective of this 2-arm parallel randomized controlled trial was to evaluate patient acceptance of the mini-implant anchored Forsus Fatigue Resistant Device (FFRD) (3M Unitek, Monrovia, Calif).
Methods
The study included 32 skeletal Class II girls. The eligibility criteria included a deficient mandible, a horizontal or neutral growth pattern, an increased overjet, and a full set of erupted permanent teeth. After the leveling and alignment stage, FFRDs and mini-implants were inserted; they were removed after the teeth reached an edge-to-edge incisor relationship. The patients were afterward asked to fill out assessment questionnaires regarding their experience with the FFRD.
Outcomes
The primary outcome of this study was to assess patient acceptance of the appliance and satisfaction with the results. The secondary outcomes were interference with functional activities, noticeability by others, pain, swelling, gum problems caused by the appliance, and appliance breakage.
Randomization
Computer random sequence generation was done using block sizes of 6 and 4. Allocation concealment was achieved with sequentially numbered opaque sealed envelopes.
Blinding
Blinding of the clinicians and the patients to the intervention was impossible, but it was done for the outcome assessment and the statistician.
Results
The 32 patients were randomly allocated in a 1:1 ratio into 2 groups: 16 patients (mean age, 13.25 ± 1.12 years) received the FFRD alone (FFRD group), and 16 patients (mean age, 13.07 ± 1.41 years) had mini-implants in conjunction with FFRDs (FMI group). No statistically significant differences were reported between the 2 groups regarding ease of appliance insertion, noticeability by others, pain, swelling, effects on eating and speech, and gum bleeding; 100% and 87.5% were satisfied with the results in the FFRD and FMI groups, respectively, with a ridit value of 0.57 (95% confidence interval, 0.43-0.71; P = 0.36). No serious harm was observed other than swelling of the cheeks, which occurred in 4 patients.
Conclusions
There were no significant differences between the patients’ acceptance of the FFRD and the mini-implant anchored FFRD. They were highly satisfied with the results. Neither group reported significant functional limitations.
Registration
This trial was not registered.
Protocol
The protocol was not published before trial commencement.
Funding
The study was self-funded by the authors.
Highlights
- •
We aimed at assessment of the patients’ acceptance to the mini- implants anchored Forsus Fatigue Resistant Device (FFRD) in comparison with the conventional FFRD.
- •
No significant differences were reported between groups regarding the ease of appliance insertion, noticeability by others, pain, swelling, effect on eating, speech, and sleep, and gum bleeding.
- •
The patients were highly satisfied with the treatment results and reported no significant functional limitations.
- •
The mini-implants anchored FFRD and the FFRD could be considered equally accepted by the patients.
In orthodontics, there is great interest in the study of patient expectations and satisfaction. The quantification of patients’ opinions can be best obtained from surveys or assessment questionnaires. The long-term nature of orthodontic treatment and the complex functional and esthetic treatment results render it a difficult process. Successful orthodontic treatment depends on patient acceptance of the orthodontic techniques and appliances used. Several factors must be considered when evaluating patient compliance with and acceptance of the various orthodontic techniques. These include personality factors, motivation, and attitudes toward orthodontic therapy, initial pain and discomfort, constraint in the oral cavity, and pressure on the soft tissues and teeth caused by the orthodontic appliances. The effect on speech and the noticeability of the appliance might have severe negative impacts on a patient’s self-confidence.
Fixed functional appliances were first introduced by Emil Herbst in 1905. They were found to be effective in the management of Class II malocclusions caused by a deficient mandible, which was the most common component of a Class II malocclusion. The appliances do not require patient compliance, and they can be used simultaneously with orthodontic brackets. The Forsus Fatigue Resistant Device (FFRD) (3M Unitek, Monrovia, Calif) was introduced by William Vogt in 2006 as a semirigid fixed functional appliance.
Most studies conducted on the FFRD have shown that the correction achieved for a Class II Division 1 malocclusion was mainly due to dentoalveolar changes that included proclination of the mandibular incisors. Recent trials were performed to minimize those side effects and to obtain skeletal effects with mini-implants as extra means of anchorage in the mandibular arch. A recent study, however, showed that no clinically significant benefit could be achieved by the addition of mini-implants to the Herbst appliance.
Bowman et al evaluated patients’ experiences with the FFRD. Gandhi et al compared patients’ attitudes toward the FFRD and the mandibular protraction appliance IV. They agreed with the claim that this appliance should have been more accepted by patients than other rigid fixed functional appliances because the patient can bite in centric occlusion and because it allows more flexible mandibular positioning. However, no studies have evaluated patients’ experiences with the mini-implant anchored FFRD. Thus, the purpose of our study was to compare patient acceptance of the FFRD when used with and without mini-implant anchorage in a sample of growing girls.
Specific objectives or hypotheses
In this study, the primary outcome was to test the hypothesis that there are no significant differences between FFRD appliances with and without mini-implants regarding patients’ acceptance and interference of the appliances with their functional activities. The secondary objectives included the equal acceptance of both groups regarding (1) satisfaction with results, (2) noticeability of the appliance by others, (3) pain, swelling, and gum problems caused by the appliance, and (4) breakage of the appliance.
Material and methods
Trial design
This parallel-group randomized controlled trial had a 1:1 allocation ratio.
Participants, eligibility criteria, and settings
The participants were recruited at the outpatient orthodontic clinic of the Faculty of Oral and Dental Medicine at Cairo University in Egypt. The study was self-funded by the authors, who were part of the university’s staff. The study method was approved by the Faculty of Oral and Dental Medicine of Cairo University’s ethical committee.
The sample comprised 32 female subjects. The criteria for the participants included the following: (1) 11 to 14 years of age; (2) skeletal and Angle Class II Division 1 malocclusion with a deficient mandible (ANB, ≥ 5°; SNB, ≤74°) as measured on pretreatment lateral cephalograms; (3) horizontal or neutral growth pattern (MMP, ≤30°) as measured on pretreatment lateral cephalograms; (4) increased overjet (minimum of 5 mm) with a Class II canine relationship (minimum of a half unit) as measured on pretreatment dental casts; and (5) a full set of erupted permanent teeth, from the first molar to the first molar of the other side, with mandibular arch crowding less than 3 mm as measured on the pretreatment dental casts.
The error of the cephamoletric measurements used for inclusion of patients was assessed by the same examiner S.A.E. after an interval of 14 days and by another examiner M.M.S.F.
Interventions
The following steps were performed for each patient.
We bonded 0.022-in slot MBT prescription brackets (3M Unitek) to both arches. The brackets on the mandibular canines and first premolars were not bonded parallel to the long axes of the teeth. They were bonded with exaggerated mesial tipping for the canines and distal tipping for the first premolars. This was done to allow for root divergence to create interradicular spaces for insertion of the mini-implants ( Fig 1 ). Leveling and alignment progressed until reaching 0.019 × 0.025-in stainless steel archwires with cinching back of the maxillary and mandibular archwires.
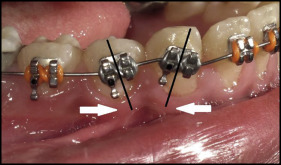
In the FMI group, mini-implants (1.6 × 10 mm; 3M Unitek) were inserted under local anesthesia between the mandibular canines and first premolars on both sides. A 0.019 × 0.025-in stainless steel wire segment was used for fixation of the mini-implants to the mandibular arch and bonded to the labial surface of the canines ( Fig 2 ).
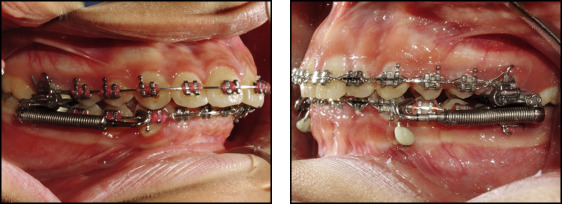
In both groups, the proper size of the FFRD was selected according to the manufacturer’s instructions and the protocol used by Franchi et al. The EZ module clip was inserted into the extraoral tubes of the maxillary first molars from the mesial to the distal sides, and the pushrods were inserted onto the mandibular archwires distal to the canines. Follow-up visits were every 3 to 4 weeks to check the mini-implants for stability and integrity of the fixation wire segment, and the appliance was checked for activation. In case of need for activation, split crimps were used according to the manufacturer’s instructions.
Treatment was continued until a Class I canine relationship was reached with overcorrection to an edge-to-edge incisor relationship ( Fig 3 ).
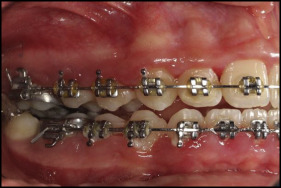
The patients of both groups were asked to fill out assessment questionnaires ( Appendix ) on the day of removal of the FFRD. The questions were thoroughly explained to the patients or the parents by the principal investigator (S.A.E.) to prevent any lack of comprehension.
Outcomes, primary and secondary, and any changes after trial commencement:
The primary outcome of this study was to assess the patients’ acceptance of the appliance and the interference of the appliance with their functional activities.
The following secondary outcomes were also assessed: (1) satisfaction with the results; (2) noticeability of the appliance by others; (3) pain, swelling, and gum problems caused by the appliance in both groups as reported by the patients; and (4) breakage of the appliance as reported by the patient and confirmed by the principal investigator (S.A.E.) from the patients’ follow-up records.
All of these outcomes were assessed using the questionnaires that were filled by the patients. The questionnaire used in our study was modified from that of Bowman et al, who investigated patients’ experiences with the FFRD. The questionnaire was designed in English and then translated into the patients’ and parents’ native language. The questionnaires were then retyped and printed to be given to the patients and their parents. Questions 1 through 11 covered the ease of appliance insertion, noticeability by others, effects on functional activities caused by the appliance, and any adverse effects of the appliance including pain and swelling. A 5-point Likert scale was used, and the questions were given scores of 0 to 4 by the patients: 0, very unsatisfied; 1, unsatisfied; 2, neutral; 3, satisfied; and 4, very satisfied. Questions 12 and 13 were yes/no questions regarding breakage of the appliances and separation of its components. Question 14 asked whether the patients would recommend this appliance to their peers.
There were no outcome changes after commencement of the trial.
Sample size calculation
Calculation of the sample size was done before the start of the study and was based on the ability to detect a minimum clinically relevant difference in the ridit scores for acceptance of the appliance regarding its interference with the functional activities in both treatment groups with an effect size of 1.2 (difference in the means of the scores of the 2 groups is 1.2 [SD, 1]). The calculation indicated that for a study with a power of 80% and an alpha error of 0.05, we needed 13 participants per group. We assumed a dropout rate of 25%; thus, a minimum of 32 patients was required. The calculation was carried out using software G* Power (version 3.1.9.2 for Windows; University of Düsseldorf, Düsseldorf, Germany).
This estimate was hypothetical because previous studies evaluating patients’ experiences with the FFRD and comparing it with other appliances used only descriptive statistics, and no data were available regarding the mean difference between groups and the standard deviations.
Interim analyses and stopping guidelines
Not applicable.
Randomization (random number generation, allocation concealment, and implementation of the random sequence)
When patients who met the inclusion criteria were identified, we asked them to take part in the trial. The patients and parents were informed about the nature of the study, and informed consents were signed.
Randomization was done by a person who was not involved in the trial. Random sequence generation was done with a computer-generated list of random numbers obtained from an Excel spreadsheet (Microsoft, Redmond, Wash) using the “=RAND()” function. To ensure a 1:1 allocation ratio, a blocked randomization was performed (random permuted blocks with randomized order of block sizes) with block sizes of 6 and 4 with the same spreadsheet. Allocation concealment was achieved with sequentially numbered and sealed opaque envelopes that were concealed from the researchers and the assessors. The envelopes contained the number that indicated the group to which the patient would be allocated and were taken by the patients after signing the informed consents. The researcher (S.A.E.) recruited the patients and explained the nature of the study to the patients and their parents using photographs of the appliances.
Blinding
Blinding of the clinicians and patients to the interventions in each group was impossible. Blinding was done for outcomes assessment, and data were extracted by an uninvolved person who did not know the nature of the intervention. The patients’ names and groups were written on a separate paper that was attached to the questionnaire. The papers with the patients’ identifications were given codes by the department librarian, who detached them from the questionnaires and stored them in an inaccessible area. Blinding of the outcomes assessment was maintained throughout the study. The person responsible for the statistical analysis was not informed about the nature of the trial. The extracted data from the questionnaires were placed in 2 tables (groups A and B) and sent to the statistician. The rows showed the values of the scores of questions 1 through 14, and the columns were given the patients’ codes. The coding was broken after all statistical tests had been performed and after the data were interpreted by the outcomes assessor as group A vs group B.
Statistical analysis (primary and secondary outcomes, subgroup analyses)
Statistical analysis was performed with SPSS software (version 20 for Windows; IBM, Armonk, NY). Descriptive statistics were performed to analyze the patients’ responses for the survey questions. Five-point Likert scale measurements were used in assessing the patients’ acceptance of various aspects of the appliances’ therapy in both groups, and a numeric value was assigned to each Likert category. The survey also included 3 yes/no questions in the descriptive statistics.
Since the data were categorical, ridit analysis was used to compare the results of the FMI and FFRD groups. Ridit analysis is a nonparametric statistical method used for comparing a sample group (in this study, the FFRD group) with a group that was defined as the reference group (in this study, the FMI group). The ridit value of the reference (FMI) group was set as 0.5. The average ridit value of the comparison (FFRD) group was calculated to make a comparison with the reference group. The latter value was interpreted as the possibility that a patient from the comparison group was in a category greater or smaller than a patient in the reference group (if it was greater or smaller than 0.5, respectively).
Error analysis for the cephalometric measurements was performed, where concordance correlation coefficients (CCCs) were calculated to detect the intraexaminer and interexaminer reliabilities of the selected cephalometric measurements used for inclusion of patients in the study. The closer the CCC value was to 1.0, the higher the reliability of the measurement. CCC values greater than 0.7 indicated good agreement, values between 0.8 and 0.9 indicated very good agreement, and values between 0.9 and 1.0 indicated excellent agreement.
Material and methods
Trial design
This parallel-group randomized controlled trial had a 1:1 allocation ratio.
Participants, eligibility criteria, and settings
The participants were recruited at the outpatient orthodontic clinic of the Faculty of Oral and Dental Medicine at Cairo University in Egypt. The study was self-funded by the authors, who were part of the university’s staff. The study method was approved by the Faculty of Oral and Dental Medicine of Cairo University’s ethical committee.
The sample comprised 32 female subjects. The criteria for the participants included the following: (1) 11 to 14 years of age; (2) skeletal and Angle Class II Division 1 malocclusion with a deficient mandible (ANB, ≥ 5°; SNB, ≤74°) as measured on pretreatment lateral cephalograms; (3) horizontal or neutral growth pattern (MMP, ≤30°) as measured on pretreatment lateral cephalograms; (4) increased overjet (minimum of 5 mm) with a Class II canine relationship (minimum of a half unit) as measured on pretreatment dental casts; and (5) a full set of erupted permanent teeth, from the first molar to the first molar of the other side, with mandibular arch crowding less than 3 mm as measured on the pretreatment dental casts.
The error of the cephamoletric measurements used for inclusion of patients was assessed by the same examiner S.A.E. after an interval of 14 days and by another examiner M.M.S.F.
Interventions
The following steps were performed for each patient.
We bonded 0.022-in slot MBT prescription brackets (3M Unitek) to both arches. The brackets on the mandibular canines and first premolars were not bonded parallel to the long axes of the teeth. They were bonded with exaggerated mesial tipping for the canines and distal tipping for the first premolars. This was done to allow for root divergence to create interradicular spaces for insertion of the mini-implants ( Fig 1 ). Leveling and alignment progressed until reaching 0.019 × 0.025-in stainless steel archwires with cinching back of the maxillary and mandibular archwires.
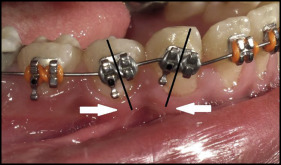
In the FMI group, mini-implants (1.6 × 10 mm; 3M Unitek) were inserted under local anesthesia between the mandibular canines and first premolars on both sides. A 0.019 × 0.025-in stainless steel wire segment was used for fixation of the mini-implants to the mandibular arch and bonded to the labial surface of the canines ( Fig 2 ).
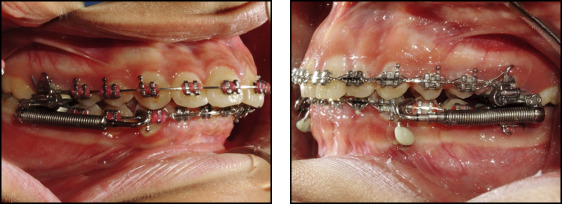
In both groups, the proper size of the FFRD was selected according to the manufacturer’s instructions and the protocol used by Franchi et al. The EZ module clip was inserted into the extraoral tubes of the maxillary first molars from the mesial to the distal sides, and the pushrods were inserted onto the mandibular archwires distal to the canines. Follow-up visits were every 3 to 4 weeks to check the mini-implants for stability and integrity of the fixation wire segment, and the appliance was checked for activation. In case of need for activation, split crimps were used according to the manufacturer’s instructions.
Treatment was continued until a Class I canine relationship was reached with overcorrection to an edge-to-edge incisor relationship ( Fig 3 ).
The patients of both groups were asked to fill out assessment questionnaires ( Appendix ) on the day of removal of the FFRD. The questions were thoroughly explained to the patients or the parents by the principal investigator (S.A.E.) to prevent any lack of comprehension.
Outcomes, primary and secondary, and any changes after trial commencement:
The primary outcome of this study was to assess the patients’ acceptance of the appliance and the interference of the appliance with their functional activities.
The following secondary outcomes were also assessed: (1) satisfaction with the results; (2) noticeability of the appliance by others; (3) pain, swelling, and gum problems caused by the appliance in both groups as reported by the patients; and (4) breakage of the appliance as reported by the patient and confirmed by the principal investigator (S.A.E.) from the patients’ follow-up records.
All of these outcomes were assessed using the questionnaires that were filled by the patients. The questionnaire used in our study was modified from that of Bowman et al, who investigated patients’ experiences with the FFRD. The questionnaire was designed in English and then translated into the patients’ and parents’ native language. The questionnaires were then retyped and printed to be given to the patients and their parents. Questions 1 through 11 covered the ease of appliance insertion, noticeability by others, effects on functional activities caused by the appliance, and any adverse effects of the appliance including pain and swelling. A 5-point Likert scale was used, and the questions were given scores of 0 to 4 by the patients: 0, very unsatisfied; 1, unsatisfied; 2, neutral; 3, satisfied; and 4, very satisfied. Questions 12 and 13 were yes/no questions regarding breakage of the appliances and separation of its components. Question 14 asked whether the patients would recommend this appliance to their peers.
There were no outcome changes after commencement of the trial.
Sample size calculation
Calculation of the sample size was done before the start of the study and was based on the ability to detect a minimum clinically relevant difference in the ridit scores for acceptance of the appliance regarding its interference with the functional activities in both treatment groups with an effect size of 1.2 (difference in the means of the scores of the 2 groups is 1.2 [SD, 1]). The calculation indicated that for a study with a power of 80% and an alpha error of 0.05, we needed 13 participants per group. We assumed a dropout rate of 25%; thus, a minimum of 32 patients was required. The calculation was carried out using software G* Power (version 3.1.9.2 for Windows; University of Düsseldorf, Düsseldorf, Germany).
This estimate was hypothetical because previous studies evaluating patients’ experiences with the FFRD and comparing it with other appliances used only descriptive statistics, and no data were available regarding the mean difference between groups and the standard deviations.
Interim analyses and stopping guidelines
Not applicable.
Randomization (random number generation, allocation concealment, and implementation of the random sequence)
When patients who met the inclusion criteria were identified, we asked them to take part in the trial. The patients and parents were informed about the nature of the study, and informed consents were signed.
Randomization was done by a person who was not involved in the trial. Random sequence generation was done with a computer-generated list of random numbers obtained from an Excel spreadsheet (Microsoft, Redmond, Wash) using the “=RAND()” function. To ensure a 1:1 allocation ratio, a blocked randomization was performed (random permuted blocks with randomized order of block sizes) with block sizes of 6 and 4 with the same spreadsheet. Allocation concealment was achieved with sequentially numbered and sealed opaque envelopes that were concealed from the researchers and the assessors. The envelopes contained the number that indicated the group to which the patient would be allocated and were taken by the patients after signing the informed consents. The researcher (S.A.E.) recruited the patients and explained the nature of the study to the patients and their parents using photographs of the appliances.
Blinding
Blinding of the clinicians and patients to the interventions in each group was impossible. Blinding was done for outcomes assessment, and data were extracted by an uninvolved person who did not know the nature of the intervention. The patients’ names and groups were written on a separate paper that was attached to the questionnaire. The papers with the patients’ identifications were given codes by the department librarian, who detached them from the questionnaires and stored them in an inaccessible area. Blinding of the outcomes assessment was maintained throughout the study. The person responsible for the statistical analysis was not informed about the nature of the trial. The extracted data from the questionnaires were placed in 2 tables (groups A and B) and sent to the statistician. The rows showed the values of the scores of questions 1 through 14, and the columns were given the patients’ codes. The coding was broken after all statistical tests had been performed and after the data were interpreted by the outcomes assessor as group A vs group B.
Statistical analysis (primary and secondary outcomes, subgroup analyses)
Statistical analysis was performed with SPSS software (version 20 for Windows; IBM, Armonk, NY). Descriptive statistics were performed to analyze the patients’ responses for the survey questions. Five-point Likert scale measurements were used in assessing the patients’ acceptance of various aspects of the appliances’ therapy in both groups, and a numeric value was assigned to each Likert category. The survey also included 3 yes/no questions in the descriptive statistics.
Since the data were categorical, ridit analysis was used to compare the results of the FMI and FFRD groups. Ridit analysis is a nonparametric statistical method used for comparing a sample group (in this study, the FFRD group) with a group that was defined as the reference group (in this study, the FMI group). The ridit value of the reference (FMI) group was set as 0.5. The average ridit value of the comparison (FFRD) group was calculated to make a comparison with the reference group. The latter value was interpreted as the possibility that a patient from the comparison group was in a category greater or smaller than a patient in the reference group (if it was greater or smaller than 0.5, respectively).
Error analysis for the cephalometric measurements was performed, where concordance correlation coefficients (CCCs) were calculated to detect the intraexaminer and interexaminer reliabilities of the selected cephalometric measurements used for inclusion of patients in the study. The closer the CCC value was to 1.0, the higher the reliability of the measurement. CCC values greater than 0.7 indicated good agreement, values between 0.8 and 0.9 indicated very good agreement, and values between 0.9 and 1.0 indicated excellent agreement.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


