Clefts of the lip and palate are the most common craniofacial birth defects, next only to congenital heart defects and clubfoot. This condition has significant psychosocial ramifications and should be managed appropriately to avoid unnecessary anxiety and exhaustion of the patient and family members. It is a treatable condition that can result in a good outcome when interdisciplinary care is provided by a team of specialists in a timely fashion from birth to adolescence. A cleft team generally includes a pediatrician, nurse practitioner, speech pathologist, pediatric dentist, orthodontist, social worker, geneticist, and surgeons with expertise in oral and maxillofacial surgery/plastic surgery and pediatric otolaryngology. The timing of various surgical interventions has been a subject of debate. This is particularly true for palate, nose, and alveolar cleft repair because of the concern about the impact of surgery on facial growth and development. The timing of surgical and nonsurgical interventions should ideally coincide with the physical, cognitive, and psychological development of the child, rather than merely chronological age. In reality, however, treatment is often dictated by socioeconomic factors, access to cleft care centers, the nutritional status of the child, and associated medical conditions. An economic evaluation of the impact of treating cleft lip and palate disorders revealed the immense gain for the individual and society. The ultimate goal of treatment is to achieve normality in speech, overall appearance, physical and psychological development, allowing the individual to be confident and successful in life. This chapter presents current evidence and rationale for timing and approaches to diagnosis and management of the child with a cleft deformity, from prenatal period to adolescence ( Box 83-1 ).
Prenatal
-
Diagnosis and parental counseling
0 to 6 Months after Birth
-
General assessment and genetics evaluation
-
Evaluation of airway, feeding, swallowing, and hearing
-
Infant presurgical orthopedics
-
Cleft lip repair
6 Months to 2 Years
-
Cleft palate repair
-
Grommets/ear tubes
-
Assess oral sensory motor development and speech
Pre-school: 3 to 5 Years
-
Evaluation of primary dentition
-
Speech assessment and therapy
-
Nasolabial revision if indicated
Childhood: 6 to 12 Years
-
Correction of velopharyngeal insufficiency
-
Orthodontic treatment (phase I)
-
Monitor facial growth and development of permanent dentition
-
Alveolar cleft repair
Adolescence: 13 to 19 Years
-
Orthodontic treatment (phase II)
-
Orthognathic surgery for correction of maxillary hypoplasia
-
Revision of secondary lip and nasal deformities
-
Replacement of missing teeth
-
Genetic counseling
Epidemiology
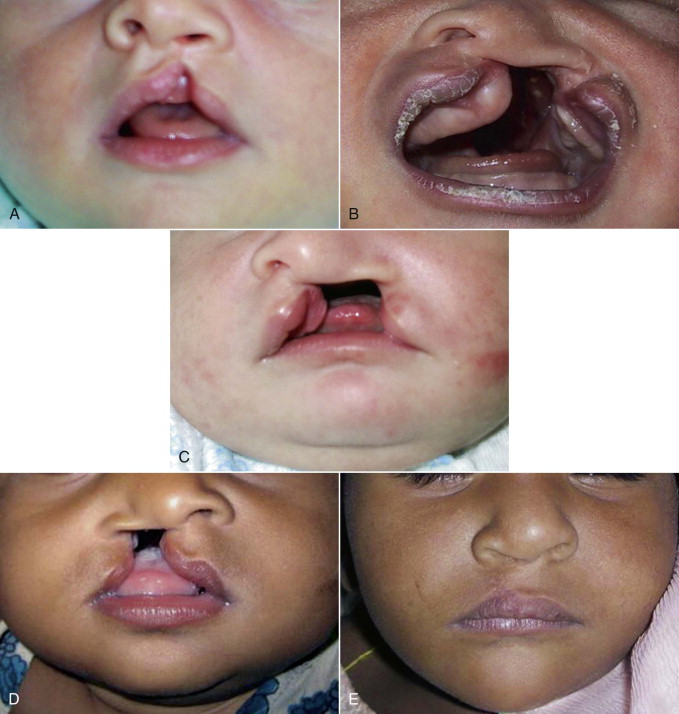
Clefts of lip and palate are the most common congenital facial defects, with an incidence ranging from 1 : 500 to 1 : 2000 live births. Cleft lip with or without cleft palate (CL/P) shows considerable variation among various ethnic groups, with Native Americans and Asians (1 : 500) clearly at a higher risk in comparison to whites (1 : 1000) and those of African descent (1 : 2000). In contrast, clefts of palate only (CP) have lower incidence (1 : 2000) and a more homogeneous distribution across all populations compared to CL/P. About half of the oral clefts involve both lip and palate (46%), a third involve only the palate (33%), and a fifth are clefts of lip alone (21%). Clefts of lip with or without palate (CL/P) are more often unilateral than bilateral and more common in males than females. The unilateral defects occur more often on the left side than right side. Clefts of lip occur in the ratio of 6 : 3:1 for unilateral left, unilateral right, and bilateral. Cleft palate, on the other hand, is more common in females and more often associated with other developmental anomalies. Clefts may be classified as syndromic or isolated depending on the presence or absence of other developmental anomalies. Syndromic association is more common in CP (50%), whereas the majority of CL/P (80%) are isolated. Oral clefts are a common presentation in many syndromes; some of the more common syndromes associated with cleft lip and palate, include Van der Woude, Down, orofacial digital, Opitz, and fetal alcohol syndrome ( Box 83-2 ). The triad of micrognathia, glossoptosis, and airway obstruction (Pierre Robin sequence) may sometimes be present in both syndromic and nonsyndromic children with cleft palate. The most common syndromic presentations of Pierre Robin sequence are Stickler’s syndrome, velocardiofacial (VCF) syndrome, and Treacher-Collins syndrome.
Distribution of Oral Clefts
-
Cleft lip and palate: 46%
-
Cleft palate only: 33%
-
Cleft lip only: 21%
Cleft Lip with or without Palate (CL/P)
-
Average birth prevalence: 1 : 700
-
More common in males
-
Unilateral > bilateral
-
Left side > right side
-
Association with other anomalies: 10%
Cleft Palate Only (CP)
-
Average birth prevalence: 1 : 2000
-
More common in females
-
Association with other anomalies: 50-60%
Some Syndromes Associated with CL/P
-
Fetal alcohol syndrome
-
Down syndrome
-
Van der Woude syndrome
-
Ectrodactyly-ectodermal dysplasia-clefting syndrome
-
Popliteal pterygium syndromes
-
Opitz syndrome
-
Craniofacial microsomia
Some Syndromes Associated with CP
-
Down syndrome
-
22q deletion syndromes: DiGeorge syndrome, Shprintzen syndrome
-
Stickler syndrome
-
Treacher Collins syndrome
-
Apert syndrome
-
De Lange syndrome
Etiology and Genetics
Nonsyndromic CL/P is a complex trait with a multifactorial etiology that results from a combination of genetic and environmental factors ( Box 83-3 ). Research to identify candidate genes and loci responsible for clefting in recent years suggests that anywhere from 3 to 14 genes contribute to cleft lip and palate. For nonsyndromic cleft lip and palate, candidate genes and loci have been identified on chromosomes 1, 2, 4, 6, 11, 14, 17, and 19. Clues from mendelian forms of clefting disorders such as Van der Woude syndrome, which is an autosomal dominant condition, have facilitated the mapping of genes responsible for nonsyndromic cleft lip and palate. Using this model, Zucchero et al. have shown that DNA-sequence variants associated with the gene IRF6 (interferon regulatory factor 6) are major contributors (12%) to CL/P. The genes IRF6 and MSX-1 and FGFR1 account for about 15% of all nonsyndromic cleft lip and palate. The genes contributing to cleft palate are different. Aberrant TGF-b3 signaling plays a role in the pathogenesis of cleft palate. Mutations in other genes TBX22, FGFR1, and P63 also contribute to syndromic clefts of the palate.
Estimated Contributions of Identified Genes to CL/P
-
IRF 6 (Interferon regulatory factor 6): 12%
-
FGFs (Fibroblast growth factors): 3%
-
MSX1(Msh homeobox 1): 2%
-
Private mutations in candidate genes: 6%
Contribution of Environmental Factors to CL/P
-
Maternal smoking
-
Maternal folate deficiency
-
Maternal diabetes
-
Maternal drug use: alcohol, steroids, phenytoin sodium
Environmental factors that contribute to the etiology of facial clefting disorders can be divided into four groups: drugs, chemicals, maternal metabolic imbalances, and maternal infections. Cigarette smoking during pregnancy, maternal exposure to alcohol and teratogenic medications such as retinoids, corticosteroids, and anticonvulsants (phenytoin and valproic acid), low dietary intake of B-complex vitamins, and folic acid deficiency during the periconceptional period can cause clefting disorders. Co-sanguinous marriages, maternal diabetes, and obesity have also been linked to an increased risk of orofacial clefts. Less consistent associations have been found between clefts and maternal viral infections such as rubella and varicella. Studies conducted to determine the risk of clefting disorders show that every parent has about a 0.14% (1 : 700) chance of having a child with CL/P and a 0.04% (1 : 2000) chance of having a child with CP. The risk of recurrence of a cleft condition is determined by a number of factors including the number of family members with clefts, their relationship to family members with clefts, race, sex, and the type and severity of cleft of the affected individuals. Studies show that the recurrence risk for first-degree relatives is about 3.3% for cleft lip with or without palate, and for isolated cleft palate it is 2%. Once parents have a child with a cleft, the risk of having a second child with a cleft is about 2-5%, and after two affected children that risk rises to 9-12%. In twins with cleft lip and palate and those with isolated cleft of palate, the concordance is far greater for monozygotic twins (43%) than for dizygotic twins (5%). Parents and young adults should be counseled appropriately by a geneticist so that they are in a better position to make decisions about future pregnancies.
Etiology and Genetics
Nonsyndromic CL/P is a complex trait with a multifactorial etiology that results from a combination of genetic and environmental factors ( Box 83-3 ). Research to identify candidate genes and loci responsible for clefting in recent years suggests that anywhere from 3 to 14 genes contribute to cleft lip and palate. For nonsyndromic cleft lip and palate, candidate genes and loci have been identified on chromosomes 1, 2, 4, 6, 11, 14, 17, and 19. Clues from mendelian forms of clefting disorders such as Van der Woude syndrome, which is an autosomal dominant condition, have facilitated the mapping of genes responsible for nonsyndromic cleft lip and palate. Using this model, Zucchero et al. have shown that DNA-sequence variants associated with the gene IRF6 (interferon regulatory factor 6) are major contributors (12%) to CL/P. The genes IRF6 and MSX-1 and FGFR1 account for about 15% of all nonsyndromic cleft lip and palate. The genes contributing to cleft palate are different. Aberrant TGF-b3 signaling plays a role in the pathogenesis of cleft palate. Mutations in other genes TBX22, FGFR1, and P63 also contribute to syndromic clefts of the palate.
Estimated Contributions of Identified Genes to CL/P
-
IRF 6 (Interferon regulatory factor 6): 12%
-
FGFs (Fibroblast growth factors): 3%
-
MSX1(Msh homeobox 1): 2%
-
Private mutations in candidate genes: 6%
Contribution of Environmental Factors to CL/P
-
Maternal smoking
-
Maternal folate deficiency
-
Maternal diabetes
-
Maternal drug use: alcohol, steroids, phenytoin sodium
Environmental factors that contribute to the etiology of facial clefting disorders can be divided into four groups: drugs, chemicals, maternal metabolic imbalances, and maternal infections. Cigarette smoking during pregnancy, maternal exposure to alcohol and teratogenic medications such as retinoids, corticosteroids, and anticonvulsants (phenytoin and valproic acid), low dietary intake of B-complex vitamins, and folic acid deficiency during the periconceptional period can cause clefting disorders. Co-sanguinous marriages, maternal diabetes, and obesity have also been linked to an increased risk of orofacial clefts. Less consistent associations have been found between clefts and maternal viral infections such as rubella and varicella. Studies conducted to determine the risk of clefting disorders show that every parent has about a 0.14% (1 : 700) chance of having a child with CL/P and a 0.04% (1 : 2000) chance of having a child with CP. The risk of recurrence of a cleft condition is determined by a number of factors including the number of family members with clefts, their relationship to family members with clefts, race, sex, and the type and severity of cleft of the affected individuals. Studies show that the recurrence risk for first-degree relatives is about 3.3% for cleft lip with or without palate, and for isolated cleft palate it is 2%. Once parents have a child with a cleft, the risk of having a second child with a cleft is about 2-5%, and after two affected children that risk rises to 9-12%. In twins with cleft lip and palate and those with isolated cleft of palate, the concordance is far greater for monozygotic twins (43%) than for dizygotic twins (5%). Parents and young adults should be counseled appropriately by a geneticist so that they are in a better position to make decisions about future pregnancies.
Embryology
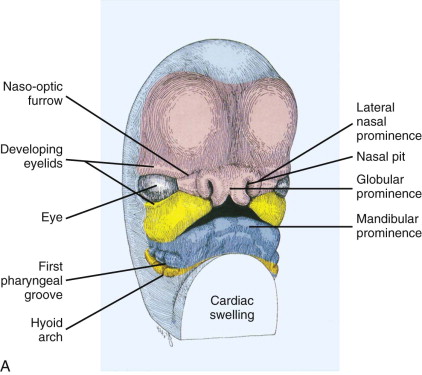
The embryo undergoes rapid changes in shape and growth between 4 and 8 weeks as the brain expands and the six branchial arches are formed. The first two branchial arches are primarily responsible for the development of the face and cranium. The development of the face begins from the neural crest ectomesenchyme that forms five prominences: the frontonasal process and paired maxillary and mandibular processes surrounding a central depression. During the 5th and 6th weeks, the bilateral maxillary processes derived from first brachial arch fuse with the medial nasal process to form the upper lip, alveolus, and primary palate. The lateral nasal process forms the alar structures of the nose. The mandibular processes form the lower lip and jaw ( Fig. 83-1 ). This process of formation of facial structures is the consequence of events such as cell proliferation, cell differentiation, cell adhesion, and apoptosis. The processes of the neural crest cells are directed by molecular signals that are controlled by an array of genes. These include fibroblast growth factors (FGFs), sonic hedgehog (SHH), bone morphogenic proteins (BMPs), members of the transforming growth factor beta (TGF-b) superfamily, and other transcription factors. Failure or error in any of these cellular mechanisms can disrupt the fusion of the medial nasal process with the lateral nasal process and maxillary process, thus causing orofacial clefts. The severity of the cleft is proportionate to the failure of penetration by the neuroectoderm.
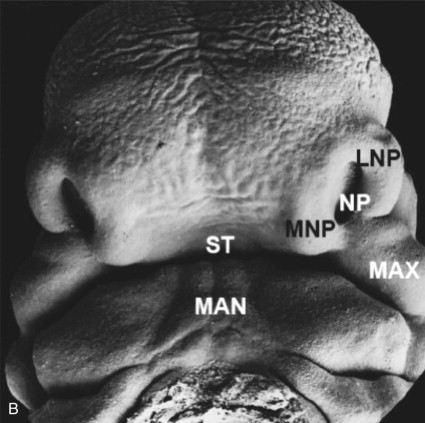
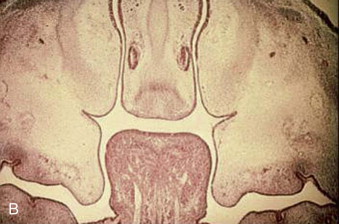
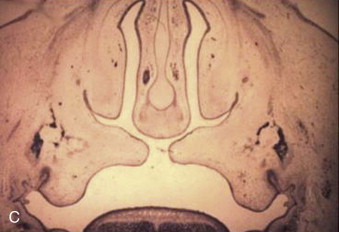
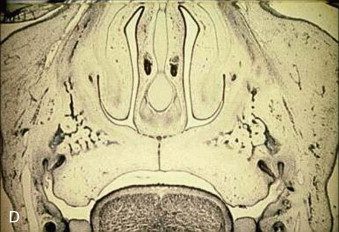
The formation of the secondary palate is embryologically different and begins during the 6th week after conception, from the two palatal shelves, which extend from the internal aspect of the maxillary processes. During the 8th week, these bilateral maxillary palatal shelves, after ascending to an appropriate position above the tongue, fuse with each other and the primary palate. A disruption in the fusion of these embryonic components can occur because of a delay in elevation of the palatal shelves from vertical to horizontal, defective shelf fusion, or postfusion rupture resulting in a cleft of the secondary palate ( Fig. 83-2 ).

Sequence of Interdisciplinary Management of Child with a Cleft Deformity
Prenatal Period
Diagnosis and Counseling
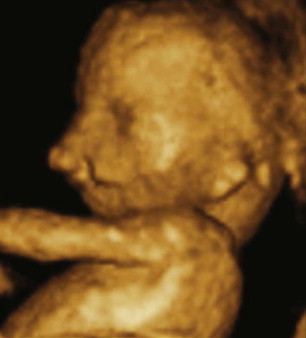
Sophisticated high-resolution 3D ultrasonography has facilitated the diagnosis of a higher percentage of craniofacial anomalies before birth ( Fig. 83-3 ). However, the majority of orofacial clefts are still detected postnatally. In the United States, about 25-30% of clefts of the lip with or without the palate are diagnosed before birth. Clefts of the lip with or without the palate can be diagnosed prenatally with a transabdominal ultrasound at 16 to 20 weeks of pregnancy and sometimes as early as 11 weeks with a transvaginal ultrasound. Clefts of the palate alone are rarely seen on ultrasound. Johnson et al. showed that the frequencies of prenatal diagnosis for cleft lip and palate, cleft lip only, and cleft palate only were 33.3%, 20.3%, and 0.3%, respectively. Several factors may influence the accuracy of ultrasound studies: the sophistication of the scanning equipment, the experience and skill of the sonographer, the number of weeks into pregnancy, the position of the baby while scanning, the amount of amniotic fluid, the maternal body structure, and the severity of the cleft. The accuracy of prenatal ultrasound diagnosis of cleft lip alone is in the range of 75-81%. Higher detection rates in some studies may be attributable to the routine inclusion of specific views of the fetal face, namely tangential and transverse, during a routine anomaly scan. The position of the fetal head (ideally occipitoposterior) enables satisfactory views in the desired planes. Premaxillary protrusion is an important clue to the presence of a complete cleft lip and cleft palate and may be more conspicuous than the cleft itself. The presence of a paranasal echogenic mass favors the presence of a bilateral cleft lip and cleft palate. A recent study showed that fetal MRI allows more detailed prenatal evaluation of the upper lip and palate than sonograms. The main advantage is that the secondary palate, which is rarely evaluated adequately on sonograms, is visible on MRI, which will enable a prospective diagnosis of clefts of secondary palate. Prenatal diagnosis and parental counseling may help families to be better prepared and provide an opportunity for possible chromosomal studies to look for other malformations. The main disadvantage of prenatal diagnosis is the anxiety, the emotional disturbance for the mother, and the inability to correct it prenatally. The capability of such tests raises both ethical and psychological issues such as the dilemma of termination of birth. Physicians and surgeons have to inform parents that cleft lip and palate in the absence of other major systemic anomalies is a treatable, non-life-threatening condition. Jones presented an algorithm for prenatal diagnosis of cleft lip and palate and strategies for counseling parents. The cleft team can discuss feeding issues, determine the timing of lip and palate surgery, and help establish contact with support groups for the family. Parents seem to be affected more by the manner in which the diagnosis is presented than the timing of the diagnosis. Although the benefits of fetal healing have been well documented, at this time there is no indication for intrauterine repair of the cleft lip deformity as the risk of fetal surgery is far too high both for the fetus and mother for correction of this non-life-threatening condition.
0 To 6 Months after Birth
General Assessment
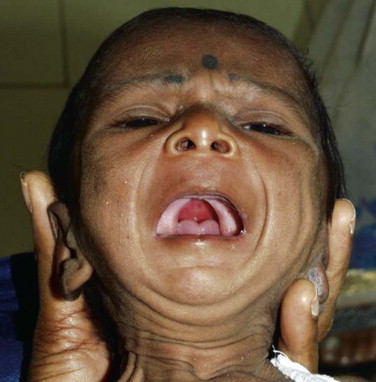
Every child born with a cleft of lip or palate should be thoroughly assessed by a neonatologist or pediatrician soon after birth ( Box 83-4 ). If the child is delivered in a nonmedical facility or a small hospital, he or she should be referred to a tertiary hospital with specialists or a craniofacial team for further evaluation. A complete physical examination including the necessary diagnostic tests should be carried out to check for airway anomalies or other associated cardiovascular, renal, and musculoskeletal abnormalities. The possibility of syndromic association should be considered, particularly in children with cleft palate ( Fig. 83-4 ). Parents should receive proper counseling for nutrition, and feeding should be initiated immediately.
- 1
Assess breathing and look for signs of airway obstruction.
- 2
Evaluate ability to feed and provide parental counseling for feeding.
- 3
Assess nutritional intake, weight gain, and growth.
- 4
Assess for concomitant anomalies (e.g., cardiac/renal/pulmonary/musculoskeletal).
- 5
Assess syndromic association and request appropriate genetic testing.
- 6
Perform craniofacial examination including head shape and circumference, ears, eyes, nose, jaws, and oral cavity.
- 7
Evaluate severity and type of cleft defect, width of cleft, position of alveolar segments and premaxilla, as well as nasal deformity.
- 8
Assess need for presurgical orthopedics and type of appliance necessary.
- 9
Prepare child and parents for surgical repair of cleft lip.

Airway Evaluation
Infants are obligate nasal breathers. Concomitant anomalies of the upper and lower airway are common in children with craniofacial birth defects. Assessment of the airway should be a priority for a newborn with a cleft deformity. Children born with a cleft of the palate may have associated micrognathia, glossoptosis, and airway obstruction ( Fig. 83-5 ). In these children, one should look for signs of increased effort while breathing, stridor, difficulty in feeding, weight loss, and failure to thrive. Parents should be informed to watch for an abnormal breathing pattern or respiratory distress, particularly during upper respiratory tract infection. If there are signs of obstruction, a pediatric otolaryngologist should be consulted to perform an endoscopic evaluation of the upper and lower airways to look for any possible cause of obstruction. In the upper airway, the patency of nares, choanae, jaw size and position, as well as tongue position in relation to the cleft of the palate, should be assessed. Examination of the lower airway to detect laryngotracheal anomalies such as tracheomalacia, laryngeal clefts, and stenosis is necessary.
Feeding and Nutrition
Children with cleft deformities have difficulty feeding because of their inability to create a proper seal around the nipple of the bottle or breast. Feeding is not a major problem when the cleft involves only the lip. Despite the problems with maintaining sucking pressures, the swallowing mechanisms in children with cleft palate are usually normal. Therefore, if the milk or formula can reach the oropharynx, the natural swallowing reflexes can move it into the esophagus. Babies with isolated clefts of the lip or palate can usually feed by mouth with some adjustments to bottle-feeding techniques. Tube feeding is rarely required. Initially, some children with cleft palate may show some discoordination of the “breathe, suck, swallow” reflex, resulting in nasal regurgitation of feeds or intermittent choking spells. These episodes often are self-limited, and the infant soon learns to prevent nasal regurgitation and coordinate swallowing. Placing the infant in an upright position during feeding may reduce the occurrence of nasal regurgitation. The strategies that have been developed to feed infants with clefts of the palate are designed to overcome the lack of negative pressure during sucking. These include, but are not limited to, cross-cut fissured feeding nipples or squeezable soft bottles that can deliver more milk under less pressure. Studies show that the use of a passive palatal feeding appliance does not improve growth outcomes of children with cleft lip and palate. The team nurse should provide the parents with information on feeding during prenatal counseling or immediately after birth. The goal is to provide adequate nutrition to satisfy the caloric requirements for growth and prepare the child for timely surgical repair of lip and palate during the first year after birth.
Hearing and Early Speech Evaluation
An audiology assessment is recommended soon after birth to check for hearing abnormalities. In children with cleft palate it is best to perform this hearing screening within the first few days of life, when the middle ear is well aerated and the child has not yet developed effusion. The screening test is most useful during this effusion-free period. These children also exhibit a higher frequency of otitis media because of eustachian tube dysfunction prior to palate repair. This can contribute to conductive hearing loss and sometimes speech and language delay. Insertion of pressure-equalizing tubes (grommets) at the time of palate repair is recommended to avoid middle ear ventilation disorders. Although not as common as conductive hearing loss, sensorineural hearing deficits exist within the cleft population, and they have an effect on speech perception and clarity, as well as auditory comprehension skills. An initial speech evaluation no later than 6 months after birth is recommended for children with cleft palate.
Presurgical Orthopedics
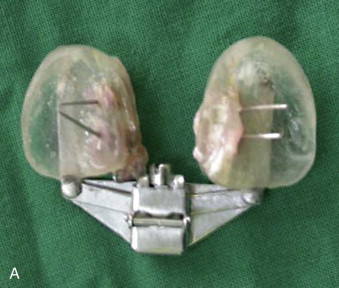
The benefits of presurgical orthopedics include better alignment of the alveolar segments, premaxilla, tension-free approximation of the cleft lip edges, and improvement of nostril symmetry as well as shape. McNeill and Burston introduced presurgical orthopedics in 1950 to align the collapsed maxillary alveolar segments prior to lip surgery using a palatal acrylic appliance. In 1975, Latham and Georgiade subsequently described the use of an active pin retained device to expand the collapsed lateral maxillary segments and retract the premaxilla in complete bilateral clefts and to achieve symmetry of the alveolar arch in complete unilateral clefts ( Fig. 83-6 ). These early techniques focused on the alveolar and premaxillary segments but did not address the deformity of the nasal cartilage.
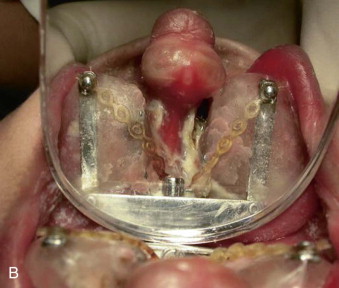
More recently, Grayson and others (1993), showed that gentle application of presurgical orthopedic forces to mold the alveolar segments and the nostrils within 3 months has shown some benefits in correction of the nasal deformity in children with complete bilateral cleft lip and palate and wide unilateral clefts. Nasoalveolar molding (NAM) increases the surface area of the nasal mucosal lining and helps to elongate and upright the columella, and improves nostril symmetry. This preoperative expansion of the nasal lining allows suturing of interdomal cartilages without tension and decreases widening of the nose. This technique also allows better position of the alveolar segments, facilitates gingivoperiosteoplasty at the time of lip repair, and may prevent the need for secondary bone grafting in select cases ( Fig. 83-7 ). In recent years, several centers have reported the adoption of the nasoalveolar molding technique to improve the outcomes of lip and nose repair, especially in complete bilateral clefts of the lip and palate, but there is a wide variation in the availability of expertise and cost of treatment. It is important to evaluate these recent studies critically for their overall clinical and cost effectiveness. Consideration should be given to other cost-effective and simple techniques of nonsurgical lip adhesion, such as lip taping ( Fig. 83-8 ) prior to surgical repair, which have been used in the past in infants with wide clefts.
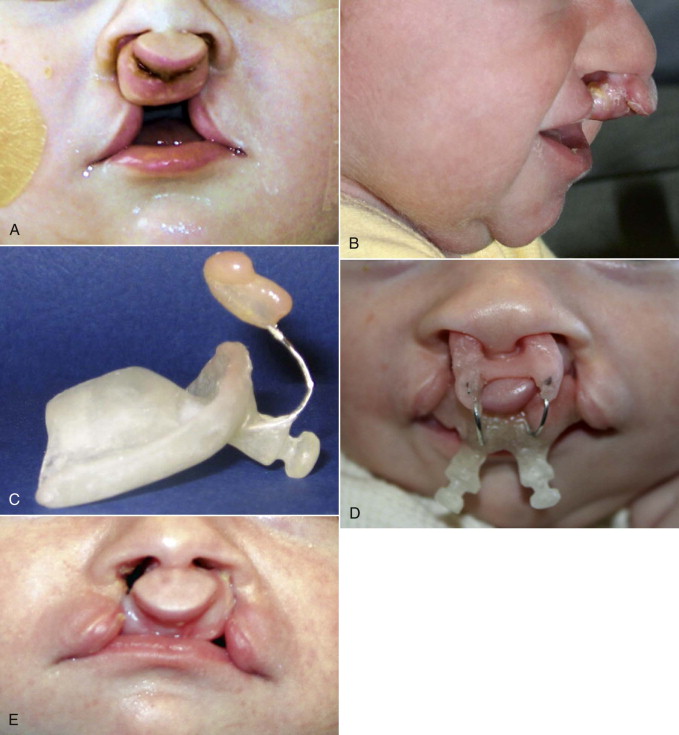
Cleft Lip Repair
Timing of Cleft Lip Repair
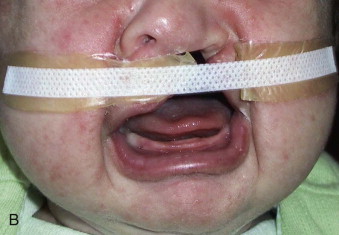
The timing for primary lip repair is usually between 3 to 6 months after birth, but it can be performed slightly earlier or later depending on the associated medical conditions and nutritional status of child. Most craniofacial centers around the world follow the rule of tens, which implies that the infant should be at least 10 weeks of age, weigh at least 10 pounds, and have at least 10 gm/100 ml of hemoglobin to withstand the surgical and anesthetic stress. In general, surgery is indicated when the infant is adequately nourished, when he or she has been thoroughly evaluated for other concomitant systemic anomalies, and when respiratory control mechanisms are mature. Ziak et al. reported that the most common reasons for delay in lip repair are prematurity, hypoxia, respiratory tract infections such as recurrent bronchopulmonary pneumonia, and anemia. In general, most centers prefer to perform the unilateral lip repair when the infant is aged 3 to 4 months. Some centers have advocated lip repair in the early neonatal period, with a theoretical benefit in the scar appearance and nasal cartilage adaptability, thus minimizing the nasal deformity. However, it appears that the main advantage in performing lip repair in this early neonatal period versus at 3 months of age is the psychological benefit and satisfaction expressed by mothers with no added benefit in terms of success of surgical outcome. By the time the baby is 3 months old, the tissue of the lip is more full and the lip elements are larger and better defined, which allows meticulous reconstruction and the child is better able to withstand the stress of surgery and anesthesia.
One Stage vs. Two-Stage Lip Repair
Primary lip repair can be staged by performing a lip adhesion initially followed by a definitive cheiloplasty. A surgical lip adhesion may be preferred as an initial surgical procedure within 6 to 8 weeks after birth as it helps to mold the alveolar segments and decreases the width of the cleft, thereby facilitating the definitive repair without tension at a later date. Good approximation of the alveolar segments also allows the surgeon to perform a gingivoperiosteoplasty at the time of definitive cheiloplasty. However, this may not be the case always, as the molded lesser segment can collapse. The disadvantages of converting the complete cleft lip to an incomplete one by lip adhesion are the need for an extra operation and the possibility of removal of more tissue at the time of definitive lip repair. Nonsurgical orthopedic techniques during the first 6 to 8 weeks after birth, as described earlier with presurgical orthopedics, can produce good alignment of the alveolar segments.
Evolution and Surgical Principles of Unilateral Cleft Lip and Nose Repair ( Box 83-5 )
Several surgeons, including Rose (1891), Thompson (1912), Blair (1930), Le Mesurier (1949), Tennison and Randall (1952), and Skoog (1974), have contributed to the evolution of cleft lip repair, but the most popular technique was introduced by Millard (1955) who described the rotation-advancement concept. Today, various modifications of the Millard advancement rotation technique, an extremely versatile procedure, are used to repair the unilateral cleft lip deformity. In Millard’s technique, the medial flap is rotated downward to achieve length, while the lateral flap is advanced. The advantage of this technique is that the suture line lies on the recreated philtral column and incision allows easy access for primary rhinoplasty to reposition the nasal septum, lower lateral cartilage, and alar base. It is a versatile technique and enables the surgeon to modify or adjust while operating, depending on the physical characteristics of the cleft. The main disadvantage is that the inexperienced surgeon requires good surgical judgment during the operation, as it is not based on exact measurements. Inadequate rotation and advancement is a common error that can produce suboptimal results. On the other hand, the triangular flap technique described by Tennison and Randall is based on exact measurements, can be reproduced well, and is used more easily in wide clefts of the lip. Despite inherent variations in different techniques, there are some similarities that form the guiding principles in surgical repair of the unilateral cleft lip deformity.
Unilateral
-
Rotation and lengthening of shortened vertical height of medial lip element
-
Advancement of tissue flap from lateral to medial
-
Alignment and approximation of orbicularis oris
-
Maintenance of Cupid’s bow and creating a philtral column on cleft side
-
Alignment of alveolar segments and restoration of continuity if possible
-
Primary repair of the collapsed alar cartilages and nasal septum
Bilateral
-
Managing the premaxilla by nasoalveolar molding prior to lip repair
-
Establishing symmetry by simultaneous bilateral repair
-
Creating appropriate design and width of prolabial flap (narrow and biconcave)
-
Reconstructing Cupid’s bow and tubercle from lateral lip elements
-
Establishing continuity of orbicularis oris muscle beneath the prolabium
-
Maintaining a vestibule between lip and alveolar segments
-
Creating columella and tip by repositioning lower lateral cartilages with interdomal sutures
-
Repositioning and cinching alar base
The basic principles and goals of unilateral cleft lip repair are to achieve symmetry, proper alignment, and continuity of the orbicularis oris muscle and creation of a philtral column on the affected side. Proper approximation of the orbicularis muscle with mattress sutures will improve the prominence of the philtral column on the cleft side. If the alveolar ridges are in a favorable position a gingivoperiosteoplasty may be performed simultaneously. In the unilateral deformity, the normal side serves as a guide to identify the key points and to plan the incisions on the cleft side. The correction of the nasal deformity at the time of primary lip repair has been well documented with long-term outcomes. It is important to mobilize the collapsed lower lateral cartilage from the overlying skin by wide dissection and undermining. The cartilage and deviated nasal septum should be anatomically repositioned and fixed with sutures to create a symmetric nostril shape ( Fig. 83-9 ).
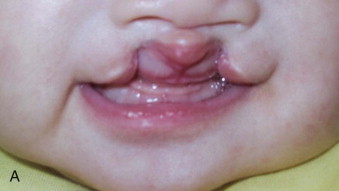
Evolution and Surgical Principles of Bilateral Cleft Lip and Nose Repair ( see Box 83-5 )
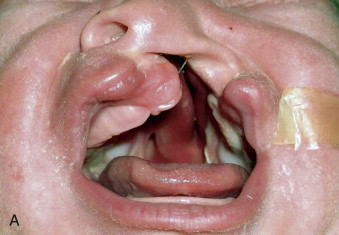
Complete bilateral clefts of lip ( Fig. 83-10 ) are rare, accounting for only 10% of cleft lips; therefore, the experience in treating these deformities is limited. Bilateral cleft lip repair is much more challenging and the results are often less satisfactory than that of unilateral cleft lip. The anatomic abnormalities that make this deformity so difficult to repair are the absence of muscle in the prolabial segment resulting in lack of philtral dimple, philtral columns, white roll margin, the median tubercle, the angular peaks, and the typical Cupid’s bow. The premaxilla is protuberant and sometimes deviated to one side, making tension-free approximation of muscle and cleft margins difficult. The orbicularis oris muscle, which is in the lateral lip elements, inserts at the alar base on each side. The accompanying nasal deformity consists of a columella that is abnormally short, a wide nasal tip, and a flared alar base as a result of the malpositioned splayed alar cartilages.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


