A review of the literature for temporomandibular disorders (TMD) has shown little appreciation for basic pain science, but with the expansion of the perspective into the broader context of orofacial pain, there is a developing interest in understanding the pathophysiology of pain as it relates to TMD and orofacial pain. The possibility of TMD being associated with neuropathic pain has received little attention.
The International Association for the Study of Pain has defined pain as “an unpleasant, sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.” This definition includes not only the sensory aspect of pain but also the emotional and interpretive or cognitive aspects of pain. The emotional factors are more significant in chronic than in acute pain and assert a significant influence that usually has to be recognized and addressed to effectively treat the patient who has chronic pain. Often, chronic pain treatment failures can be traced to ignoring the psychologic issues that are affecting the patient’s pain condition.
The understanding of chronic pain has advanced significantly in the last 10 years. This understanding has led to improved diagnosis and treatment strategies for pain. Until recently, patients who had facial pain that did not fit the existing understanding and taxonomy were given the diagnosis of “atypical facial pain.” The recent IHS Classification of Headache provides a comprehensive classification system for head and neck pain and has removed the “atypical facial pain” diagnosis in favor of “persistent idiopathic facial pain.” This is an important step in disengaging the less understood facial pain condition from a co-psychosomatic diagnosis that was implied in atypical facial pain .
To be able to diagnose and treat orofacial pain, one must understand basic neurophysiology of pain from the periphery to the central nervous system (CNS). This article describes the basis of central sensitization as it relates to orofacial pain.
Pain transmission from periphery to central nervous system
Afferent sensory system: C-polymodal nociceptors and A-δ and A-β fibers
A basic understanding of the peripheral and CNS is necessary to understand pain mechanisms and to understand how central sensitization develops. Most text books on pain discuss dorsal horn mechanisms when referring to the CNS. For orofacial pain, the trigeminal correlate of the dorsal horn is the trigeminal nucleus within the pontine brain stem. Peripherally, the trigeminal nerve provides sensory input from the anterior part of the head, including the intraoral structures. Because the nociceptive endings of pain fibers lack specialized receptors, they are named from their afferent fiber and the stimulus that activates them. The sensory fibers are divided into A-β mechanoreceptors and three types of nociceptors: A-δ fibers, C–polymodal nociceptors (C-PMNs), and silent or sleeping nociceptors, which are unmyelinated or thinly myelinated. The A-β fibers that respond to light-touch mechanostimulation are large diameter, fast conducting, and myelinated. No matter what the frequency or intensity of the stimulus, these fibers normally encode only low-frequency, non-noxious stimuli that are interpreted as light touch . After trauma, the A-β fibers may begin to signal pain. The A-δ fibers respond to painful mechanical stimuli with an output in the high-frequency range. This is perceived as sharp or stabbing pain. Because the A-δ fibers are myelinated, the convey impulses more rapidly than the C-PMNs ( Fig. 1 ) . The silent nociceptors are normally mechanically insensitive. They become active when tissue is injured. These fibers add to the nociceptive input to the CNS . The afferent impulses from all the sensory fibers travel from the periphery through the trigeminal ganglion and trigeminal root, enter the pons, and descend in the trigeminal tract to enter the trigeminal nucleus. Once the fibers have entered the pons, they are in the CNS.
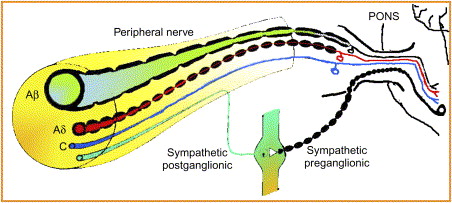
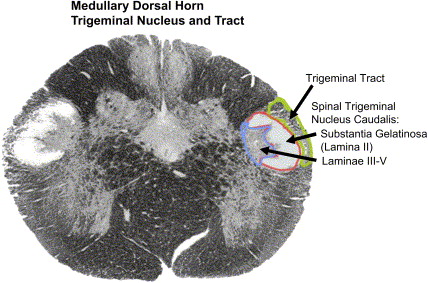
The trigeminal nerve innervates the anterior of the head. These fibers travel to the trigeminal ganglion and to the trigeminal nucleus in the pons. The trigeminal nucleus is subdivided into three parts: the uppermost subnucleus oralis, the middle subnucleus interpolaris, and the subnucleus caudalis ( Fig. 2 ) . Most of the pain fibers synapse in the subnucleus caudalis. For pain, the wide dynamic range neurons (WDRs) are the most important second-order neurons in the subnucleus caudalis. They receive convergent sensory input from primary afferent nociceptors and low-threshold mechanoreceptors.
Certain features of pain have long puzzled clinicians and researchers. The stimulation of pain from a normally nonpainful stimulus has defied explanation. Conversely, Beecher puzzled over a battlefield phenomenon he noted during the Second World War on Enzio Beach in Italy. Beecher attracted attention to the role of cognitive appraisal with his observations that soldiers wounded during battle complain far less than civilians comparably injured during accidents, presumably because the soldiers were relieved that they had escaped from the battlefield and expected to return home, whereas the civilians evaluated the injury as a threat to comfortable, established lives. Contrasting findings have shown that people who “catastrophize” or self-alarm by focusing negatively upon their distress suffer higher levels of anxiety and are the most disabled and benefit the least from conventional medical care . Patients who have chronic low back pain and are depressed have also been found to misinterpret or distort the nature and significance of their pain. These observations highlight the presence of pain-modulating systems in the body that can turn down or turn up the volume control for pain. This had been implied by Melzack and Wall but was poorly understood when they proposed the Gate Control Theory in 1965.
Second-order neurons
The first interface between the peripheral nociceptors and the CNS occurs in the spinal cord or trigeminal nucleus, the brainstem extension of the spinal cord dorsal horn (see Fig. 2 ). There are many types of receptors and ion channels associated with the cell membrane of the WDR that modulate cell activity. Modulatory circuits can suppress WDR activity and decrease pain or facilitate pain transmission.
The Gate Control Theory and pain modulation
Fig. 3 shows the Gate Control of Pain that was proposed by Melzack and Wall in 1962 and republished in 1965. Although there have been some modifications to the original theory, most of the system features have been confirmed by research.
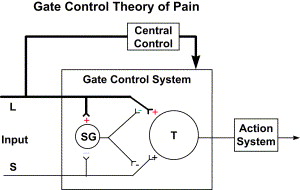
The Melzack and Wall model describes modulation of pain transmission through the interneuron connections in the substantia gelatinosa. Past research had identified a pain-modulating effect of afferent activity from large-diameter A-β fibers. The gate control model identified the spinal cord substantia gelatinosa as one of the areas where pain is modulated. Fig. 3 illustrates the modulating effect of the L (light touch fibers) in reducing the effect of afferent activity from the S (c-nociceptors) fibers. Melzack and Wall also theorized that there were descending inhibitory and facilitatory influences, but little was known of these mechanisms in 1965, and it has only been within the last few years that descending inhibitory and facilitatory systems have been identified.
The Gate Control Theory and pain modulation
Fig. 3 shows the Gate Control of Pain that was proposed by Melzack and Wall in 1962 and republished in 1965. Although there have been some modifications to the original theory, most of the system features have been confirmed by research.
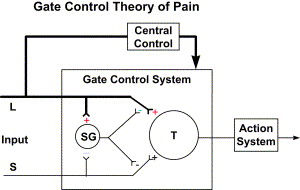
The Melzack and Wall model describes modulation of pain transmission through the interneuron connections in the substantia gelatinosa. Past research had identified a pain-modulating effect of afferent activity from large-diameter A-β fibers. The gate control model identified the spinal cord substantia gelatinosa as one of the areas where pain is modulated. Fig. 3 illustrates the modulating effect of the L (light touch fibers) in reducing the effect of afferent activity from the S (c-nociceptors) fibers. Melzack and Wall also theorized that there were descending inhibitory and facilitatory influences, but little was known of these mechanisms in 1965, and it has only been within the last few years that descending inhibitory and facilitatory systems have been identified.
Central pain processing and central sensitization
The phenomenon of peripheral sensitization develops from an injury-induced inflammatory response. Allodynia and hyperalgesia in this model are due to the inflammatory mediators being released at the site of injury. In a tooth extraction site, the inflamed area is marked by increased sensitivity to pressure (static hyperalgesia) that is mediated by sensitized nociceptors. It is expected that this reaction will resolve within a reasonable period of time due to the decreasing activity of the nociceptors and consequent decrease in afferent activity to the dorsal horn. If the inflammatory process and consequent afferent activity is of sufficient intensity and if there has been neuronal damage, a central process is established that increases sensitization, lowers the threshold of response, and causes ectopic discharges (physiologic changes). Additionally, A-β fibers begin signaling pain (dynamic mechanical allodynia), and their inhibitory effect is lost (anatomic changes and disinhibition). There is now an increased central release of excitatory mediators, such as glutamate and nitric oxide production (neurochemical changes). These changes stimulate the MAP kinase cascades, resulting in messenger RNA–mediated changes that alter the phenotype of nociceptors and mechanoreceptors such that normal cell response becomes genetically changed to a pathologic state ( Fig. 4 ).
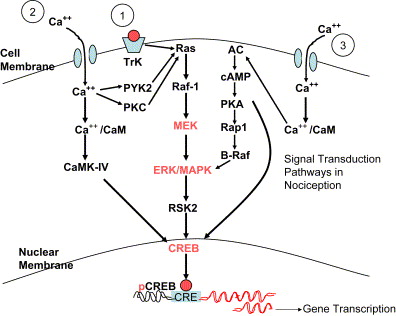
Central sensitization is a form of neuroplasticity in which nociceptor activity triggers a prolonged increase in the excitability of dorsal horn neurons. It is initiated by a brief burst of C-fiber activity. The peripheral manifestation of this central process is dynamic hyperalgesia. Torebjork has provided evidence showing that once central sensitization has occurred, Aβ fiber afferents begin to evoke painful response (allodynia) . C-nociceptors have been identified as the primary nociceptor involved in the initiation of central sensitization due to the slow synaptic currents they generate and the low-stimuli repetition rates that cause an increased rate of depolarization in the dorsal horn . This occurs as a result of the activation of ligand-gated ion channels, initially the alpha-amino-3-hydroxy-5-methyl-4-isoxazole proprionate (AMPA) receptor allowing calcium to enter the cell through the calcium channels. In addition, activation of the metabotropic glutamate and neurokinin receptors by glutamate and substance P causes a G-protein–coupled transduction signal that releases calcium from intracellular stores, further increasing the intracellular calcium levels. This calcium activates a calcium-dependent enzyme system, including protein kinases that phosphorylate the N-methyl-D-aspartate (NMDA) receptor. The NMDA receptor at normal resting membrane potentials has a magnesium ion block in the channel, but when the receptor is phosphorylated, the ion is released. Before phosphorylation, the NMDA receptor generates little inward current when glutamate is bound, but after phosphorylation and release of the ion channel block, the NMDA receptor generates inward synaptic currents at normal resting membrane potentials . This process causes increased glutamate sensitivity and is the underlying mechanism that is represented by the expansion of receptive fields and a decrease in the threshold of the dorsal horn neurons.
Aβ fiber–mediated dynamic hyperalgesia may also be the result of central reorganization of neuronal connections in the dorsal horn. Woolf and others have found that A-β fibers sprout into dorsal horn lamina I and II after peripheral injury, forming new connections in areas normally occupied only by c-fiber nociceptors. These new connections can apparently signal pain. Additionally, it has been reported that with the neuronal organization and transcriptional changes induced by the sensitization, Aβ fibers begin expressing substance P, previously thought to be associated only with c-fibers . μ−Opioid receptors are found presynaptically on c-fibers but not on A-β fibers. Part of the descending inhibitory system uses endogenous opioid action on presynaptic μ-opioid receptor. Because these receptors are not found on A-β fibers, this may account for the relative lack of response to opioid agonists in neuropathic pain.
The influx of calcium through voltage-gated ion channels also occurs on the inhibitory interneurons in lamina II. Calcium may induce excitotoxic cell death, resulting in a loss of inhibitory connections . Mao and colleagues showed that pretreatment with NMDA receptor antagonists seemed to protect the dorsal horn from changes that produced prolonged sensory hypersensitivity. Nitric oxide, arachidonic acid, superoxide, and intracellular calcium overload are the ultimate mediators of neuronal death.
Pain-modulating circuits
Pain is strongly affected by emotions. In the presence of anger, fear, or elation, major injury may be essentially painless. Conversely, in situations associated with dysphoria or when pain is anticipated, subjects often report the occurrence or worsening of pain without additional noxious stimulation. Psychologic factors influence the firing of dorsal horn pain transmission neurons.
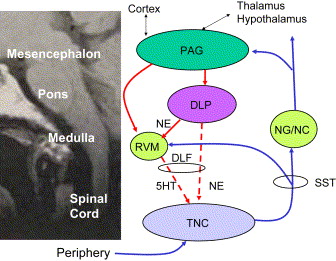
It has been observed that stimulation of the periaquaductal gray area in the midbrain increased tail-flick latency in rats that were given a painful stimulus. The periaquaductal gray area was demonstrated to be heavily innervated with serotonergic neurons. Subsequently it has been demonstrated that there are connections to the nucleus raphe magnus of the rostral ventral medulla and thence to the nucleus caudalis of the trigeminal nucleus or the dorsal horn of the spinal cord. This system is part of the descending inhibitory system mediated by serotonin. Additionally, a descending system modulated by norepinephrine travels from cortical stimulatory centers to the periaquaductal gray and on to the dorsolateral pontine tegmentum area of the medulla, also connecting to the relay neurons (wide dynamic range) in the nucleus caudalis or dorsal horn. The dorsolateral pontine tegmentum is directly linked to the periaquaductal gray and rostral ventral medulla and projects directly to the spinal cord dorsal horn and the nucleus caudalis. Pain modulation requires action from both circuits acting in tandem ( Fig. 5 ).