Fig. 14.1
Structure and synthesis of Aa leukotoxin. (a) Polypeptide domain structure. The polypeptide is 1,050 amino acid residues in length. The pore-forming region is the large N-terminal domain (dark green; residues 5–654) plus the short C-terminal domain (light green; residues 757–774). The large N-terminal domain is followed by a region that is common to all RTX proteins. It consists of two domains, one multicolored (yellow, red, and blue; residues 660–695) and one colored blue only (residues 696–720) and a tandem repeat sequence (red; residues 721–846) from which the protein family takes its name (see text). Each repeat contains the Ca2+ ion binding consensus sequence: GGXGXDXФX where Ф is a hydrophobic residue and X is any amino acid (see text). (b) Synthesis and secretion. The lktCABD operon is responsible for the synthesis, maturation, and secretion of Aa leukotoxin (protein A; green). The promoter sequence (p) is shown along with an operon polarity suppressor sequence (ops) and a termination suppressor protein (RfaH) that binds to the ops nucleotide sequence and facilitates expression of the downstream proteins B (white) and D (yellow). Proteins B and D are expressed at lower levels due to transcription termination within the operon and are required to secrete the leukotoxin. Prior to secretion, a lysine residue near the C-terminal end of the pore-forming domain (possibly lys 555, which corresponds to lys 565 in the homologous E. coli hemolysin) is acylated with an unknown fatty acid. A second acylation site (lys 691 in the E. coli hemolysin) is absent from Aa leukotoxin. Acylation precedes secretion into the extracellular medium, after which Ca2+ ion binding to the leukotoxin repeat sequences activates the toxin. TdeA is an outer membrane protein homologous to TolC and encoded by a gene outside the lktCABD operon (see text) (a: From http://pfam.sanger.ac.uk/protein?acc=P16462; b: Adapted from Fig. 1 in Stanley P, Koronakis V, and Hughes C (1998) “Acylation of Escherichia coli hemolysin: A unique protein lipidation mechanism underlying toxin function.” Microbiology and Molecular Biology Reviews 62(2):309–333. Modified for Aa as described by Crosby JA and Kachlany SC (2007) “TdeA, a TolC-like protein required for toxin and drug export in Aggregatibacter (Actinobacillus) actinomycetemcomitans.” Gene 388:83–92. With permission)
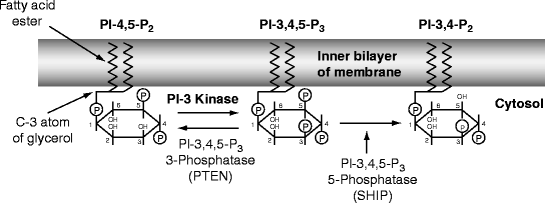
Fig. 14.2
Mode of action of cytolethal distending toxin. Phosphatidyl-inositol is attached to the inner layer of a cell membrane by its fatty acids. Inositol is a six-membered carbon ring with an OH group on each carbon atom. The atoms of the inositol ring are numbered counter-clockwise from the C1 atom, which is attached to a membrane diacylglycerol phosphate by the C3 atom of the latter’s glycerol moiety. The C2 and C1 atoms are attached to fatty acids. Inositol phosphate is phosphorylated by membrane kinases at the -4 and -5 positions and then the -3 position to make phosphatidyl inositol-3,4,5-trisphosphate (PI-3,4,5-P3), which activates growth and various biological processes. In most cells, production of PI-3,4,5-P3 is kept low by the enzyme PTEN (see text), but in macrophages SHIP is activated to remove the 5-phosphate (see text). Cytolethal distending toxin (Cdt) is a homologue of SHIP and therefore competes for PI-3,4,5-P3 only in activated leukocytes; other cells are less sensitive (see text) (Modified from Fig. 1 (p. 1171) in Sly LM, Rauh MJ, Kalesnikoff J, et al. (2003) “SHIP, SHIP2, and PTEN activities are regulated in vivo by modulation of their protein levels: SHIP is up-regulated in macrophages and mast cells by lipopolysaccharide.” Experimental Hematology 31:1170–1181. With permission) Edited by Foxit Ready Copyright (c) by foxit software company, 2005–2008. for Evaluation only.
One form of generalized aggressive periodontitis is Acute Necrotizing Ulcerative Gingivitis (ANUG), clearly a misnomer. The junctional epithelial attachment with its underlying cells and collagen fibers is rapidly destroyed along with coronal alveolar bone (see definition of coronal in Sect. 3.1.5). The amount of attachment lost in 10 days may take many years to occur in chronic periodontitis.
A key feature of ANUG is necrosis of sulcular and junctional epithelial cells and of fibroblasts and osteoblasts in the underlying stroma. Alveolar bone may be exposed in the oral cavity and the resulting pain usually leads a patient to seek help. In necrosis, the plasma membrane ruptures (Sect. 3.4.1), causing a release of apoptotic and inflammatory agents into the extracellular medium. TRAIL and FAS mediate extrinsic apoptosis of adjacent cells (Sect. 13.4.2) and TNFα and IL-1 mediate a powerful release of pro-inflammatory cytokines from more distant cells. The exact cause of ANUG is unknown, but it usually appears in individuals who have been severely stressed. Hormonal responses to severe stress alter the junctional epithelial cells response to TLR or NOD receptor activation by PAMPs from the successor microbiota (Sect. 13.2.1).
During ANUG, acid and lysosomal enzymes are released into the extracellular environment and acid activated. Cathepsin L hydrolyzes uncalcified collagen fibers, and cathepsin K hydrolyzes calcified collagen fibers due to osteoclast activation. Cathepsin L is homologous to cathepsin K but has a different specificity. In necrotic tissues, cathepsin L cleaves the nonhelical telopeptides regions of types I and II collagen. These collagen peptides are absorbed and passed to the bloodstream where they provide a unique blood plasma marker for necrotic bone destruction (Sect. 4.2.2). Cathepsin L is also involved in the macrophage digestion (processing) of foreign material (antigens) during the acquisition of immunity.
Summary
Aggressive periodontitis occurs in less than 0.1% of the US population. ANUG is a form of aggressive periodontitis in which stress hormones may overactivate TLRs to PAMPs around and within cells at the base of gingival sulci. The junctional epithelium fragments and its cells release their contents (necrosis
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


