CHAPTER 9. Non-odontogenic tumours of the jaws
Almost any type of tumour can arise in the jaws, but most of them are considerably more common in other parts of the skeleton. Only the more important examples (Box 9.1) are considered here.
Box 9.1
Non-odontogenic tumours of bone
Primary – benign
• Osteoma
• Osteochondroma
• Cemento-ossifying fibroma
• Central giant cell granuloma
• Haemangioma
• Melanotic neuroectodermal tumour
Primary – malignant
• Osteosarcoma
• Chondrosarcoma
• Ewing’s sarcoma
• Multifocal or potentially multifocal:
Myeloma
Langerhans cell histiocytosis
Metastatic
• Carcinoma
OSTEOMA AND OTHER BONY OVERGROWTHS → Summary p. 172
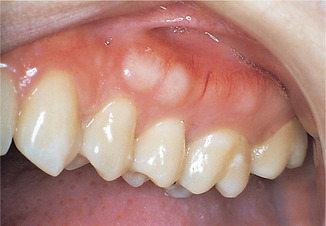
True tumours consisting of bone (either compact or cancellous) are occasionally seen, but localised overgrowths of bone (exostoses) are more common. They consist of lamellae of compact bone, but large specimens may have a core of cancellous bone. Small exostoses may form irregularly on the surface of the alveolar processes (Fig. 9.1) and specific variants are torus palatinus and torus mandibularis. They differ from other exostoses only in that they develop in characteristic sites and are symmetrical.
 |
| Fig. 9.1
Exostoses. Bony exostoses, aside from tori, are found most frequently buccally on the alveolar bone and are often symmetrically arranged.
|
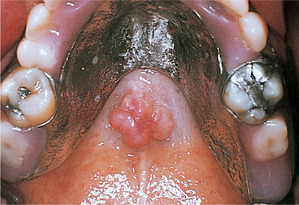
Torus palatinus commonly forms towards the posterior of the midline of the hard palate (Fig. 9.2). The swelling is rounded and symmetrical, sometimes with a midline groove. It is not usually noticed until middle age and, if it interferes with the fitting of a denture, should be removed.
 |
| Fig. 9.2
Torus palatinus. Palatal tori range from small smooth elevations to lobular swellings such as this. The bone is covered by only a thin mucosa which is prone to trauma.
|
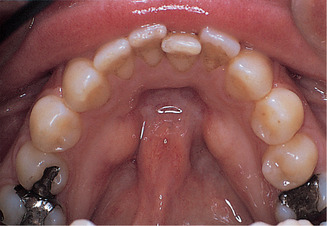
Tori mandibularis form on the lingual aspect of the mandible opposite the mental foramen. They are typically bilateral, forming hard, rounded swellings (Fig. 9.3). The management is the same as that of torus palatinus.
 |
| Fig. 9.3
Mandibular tori. The typical appearance of bilateral tori lingual to the lower premolars.
|
Compact and cancellous osteoma
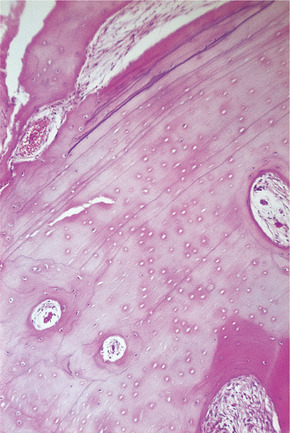
Compact osteomas consist of lamellae of bone, sometimes in layers like an onion but not in Haversian systems (Fig. 9.4). This dense bone contains occasional vascular spaces and grows very slowly. Cancellous osteomas consist of slender trabeculae of bone, with interstitial marrow spaces and a lamellated cortex (Fig. 9.5).
 |
| Fig. 9.4
Compact osteoma. Dense bone is laid down in lamellae with occasional vascular spaces, but there is no attempt to form Haversian systems.
|
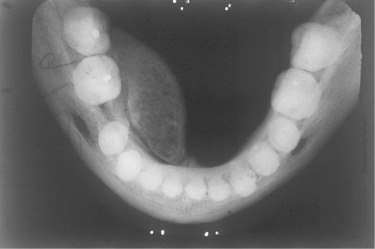 |
| Fig. 9.5
Cancellous osteoma of the mandible. The tumour has arisen from a relatively narrow base lingually to the molars but has been moulded forward during growth by pressure of the tongue. The trabecular pattern of cancellous bone can be seen. A torus mandibularis arises further forward on the jaw, arises from a broad base and is usually bilateral.
|
Osteomas should be excised only if they become large enough to cause symptoms or interfere with the fitting of a denture.
Gardner’s syndrome
Gardner’s syndrome comprises multiple osteomas of the jaw, polyposis coli with a high malignant potential and often other abnormalities such as dental defects and epidermal cysts or fibromas. It is inherited as an autosomal dominant trait but penetrance is weak.
Early recognition of these oral features should prompt bowel radiography or endoscopy and possibly prophylactic colectomy.
OSTEOCHONDROMA (CARTILAGE-CAPPED OSTEOMA)
These bony overgrowths grow by ossification beneath a cartilaginous cap. Most arise from the region of the coronoid or condylar process and form a hard bony protuberance which can interfere with joint function. The cartilaginous cap may not be visible in radiographs. Almost any age can be affected.
Pathology
The lesion is subperiosteal and has a cap of hyaline cartilage where the cartilage cells are sometimes regularly aligned or irregular and contain minute foci of ossification (Fig. 9.6). As age advances, the mass progressively ossifies.
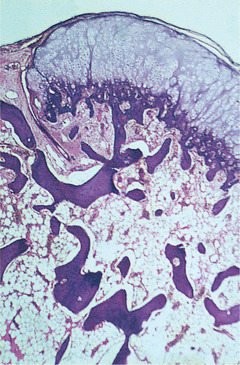 |
| Fig. 9.6
Osteochondroma. There is a superficial cap of hyaline cartilage which undergoes endochondral ossification to form normal trabeculae of lamellar bone. The marrow spaces contain normal marrow continuous with those in the underlying bone.
|
These tumours are benign and usually cease to grow after skeletal maturation. Removal is therefore curative.
CEMENTO-OSSIFYING FIBROMA → Summary p. 157
Cemento-ossifying fibroma has been discussed in Chapter 8, as its presence in the jaw and cemental content suggest an odontogenic origin.
GIANT CELL GRANULOMA → Summary p. 136
The giant cell granuloma is a localised tumour of fibrous tissue containing numerous osteoclasts. It is not related to the giant cell tumour (osteoclastoma), which by contrast is an aggressive neoplasm of long bones.
Pathogenesis
The earlier name ‘giant cell reparative granuloma’ derives from the unconvincing idea (also applied to solitary bone cysts) that it was a reaction to trauma which caused intramedullary haemorrhage. The cause is unknown though they are generally considered hyperplastic or reactive rather than neoplastic. Identical lesions occur in Noonan syndrome, the genetic cause of which is known to be mutation of the PTPN11 gene and a chromosomal translocation t(X;4) has been identified in a long bone lesion.
Giant cell granuloma is usually seen in young people under 20 and in females twice as frequently as males. The mandible, anterior to the first molars, where the teeth have had deciduous predecessors, is the usual site. There is frequently only a painless swelling, but growth is sometimes rapid and the mass can, rarely, erode through the bone, particularly of the alveolar ridge, to produce a purplish soft-tissue swelling (Fig. 9.7).
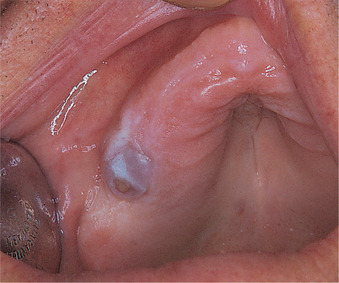 |
| Fig. 9.7
Giant cell granuloma. This maxillary lesion has perforated the cortex and formed an ulcerated bluish soft-tissue mass on the alveolar ridge. The underlying alveolar bone is considerably expanded.
|
Radiographs show a rounded cyst-like radiolucent area, often faintly loculated or with a soap-bubble appearance (Fig. 9.8). Roots of teeth can be displaced or occasionally resorbed.
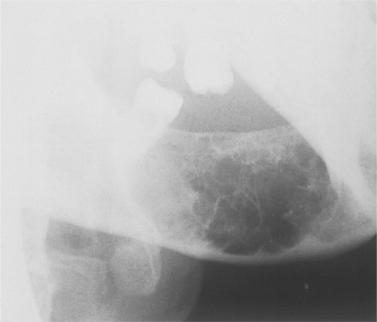 |
| Fig. 9.8
Giant cell granuloma. Characteristic radiographic appearance of an expansile radiolucency with scalloped margins containing numerous thin internal septa of wispy bone and osteoid.
|
Pathology and management
Giant cell granuloma forms a lobulated mass of proliferating vascular connective tissue packed with giant cells (osteoclasts) (Figs 9.9 and 9.10). Signs of bleeding into the mass and deposits of haemosiderin are frequently seen. Fibroblastic proliferation or prominent osteoid and bone formation are common. There are no changes in blood chemistry.
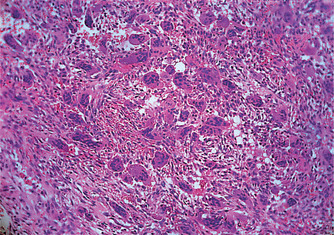 |
| Fig. 9.9
Giant cell granuloma. This tissue is vascular and contains much extravasated blood, around which giant cells are clustered.
|
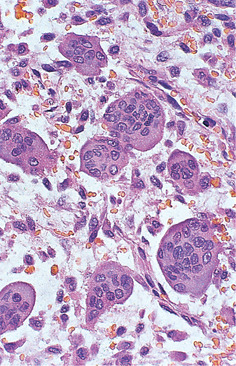 |
| Fig. 9.10
Giant cell granuloma. High power showing the osteoclast-like giant cells with numerous, evenly dispersed nuclei.
|
Curettage of giant cell granulomas is usually adequate and wide excision unnecessary; small fragments that may be left behind may cause little trouble, rarely require further treatment and may resolve spontaneously. Recurrence follows incomplete removal and a further limited operation may become necessary.
Box 9.2
Central giant cell granuloma: key features
• Benign hyperplastic lesion of unknown aetiology
• More common in young females but seen over a wide age range
• Very expansile and may be destructive. May penetrate cortical bone and periosteum
• Solid but appears as unilocular or multilocular cyst-like radiolucency
• Forms in alveolar ridge, anterior to 6s, more frequently in the mandible but often in the maxilla
• No changes in blood chemistry
• Treated by curettage but second operation sometimes required
Other giant cell lesions of the jaws
Several other jaw lesions (Box 9.3) can resemble a giant cell granuloma microscopically and must be considered in the differential diagnosis.
Box 9.3
Differential diagnosis of giant cell lesions of the jaws
• Hyperparathyroidism. Histologically indistinguishable from giant cell granuloma but serum calcium levels are raised (Ch. 10)
• Cherubism. May be indistinguishable from giant cell granuloma histologically, but lesions are symmetrical, near the angles of the mandible (Ch. 10)
• Giant cell tumour (osteoclastoma). Aggressive tumour of long bones. Broadly similar histologically to giant cell granuloma but a distinct entity in terms of behaviour
• Aneurysmal bone cysts may contain many giant cells but consist predominantly of multiple blood-filled spaces (Ch. 10)
• Fibrous dysplasia. Only limited foci of giant cells. No defined margins radiographically. Growth ceases with skeletal maturity (Ch. 10)
HAEMANGIOMA OF BONE → Summary p. 135
Haemangiomas are rare tumours of bone, but a relatively high proportion are in the mandible, particularly in women.
Clinically, haemangiomas cause progressive painless swellings which, when the overlying bone is resorbed, may become pulsatile. Teeth may be loosened and there may be bleeding, particularly from the gingival margins involved by the tumour.
Radiographically, the features of haemangioma are extremely varied. They range from a sharply defined cyst-like appearance to poorly defined pseudoloculated radiolucencies or a soap-bubble appearance. A serpiginous outline is particularly suggestive.
Pathology
Haemangiomas of bone are essentially similar to those in soft tissues. They are usually cavernous (Fig. 9.11), but there is also an arteriovenous type (fast-flow angioma) which has large feeder arteries, tends to expand rapidly and is likely to bleed severely if opened.
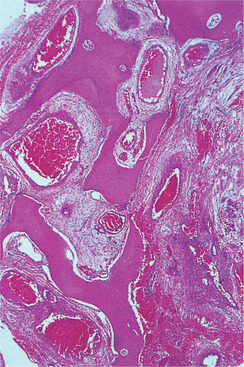 |
| Fig. 9.11
Haemangioma of bone. The marrow spaces contain very large blood-filled sinuses with thin walls; the lesion is poorly localised and permeates between the bony trabeculae.
|
Management
A haemangioma may not be suspected if its vascularity is not obvious. Opening it or extracting a related tooth may therefore release torrents of blood, occasionally with fatal results. However, once the diagnosis has been made, wide en bloc resection is the only practical treatment. If there are identifiable feeder vessels selective arterial embolisation makes surgery considerably safer and may be all that is required.
MELANOTIC NEUROECTODERMAL TUMOUR OF INFANCY (PROGONOMA) → Summary p. 337
This rare tumour, which arises from the neural crest, may appear in the anterior maxilla in the first few months of life. It is usually painless and slowly expansive, but occasionally grows rapidly.
Radiographically, there is an area of bone destruction, frequently with ragged margins, and displacement of the developing teeth (Fig. 9.12).
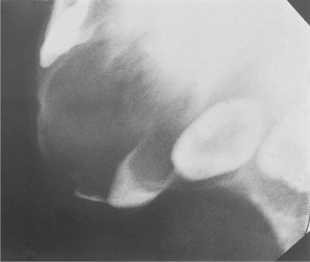 |
| Fig. 9.12
Melanotic neuroectodermal tumour of infancy. The anterior maxilla of this neonate contains an expansile radiolucency which has displaced tooth germs and eroded the alveolar bone.
|
Genetics
A molecular genetic study has shown chromosomal deletions at 1p together with translocations t(11;22) in a small number of cases. What genes are affected by these chromosomal anomalies is unknown. However, these changes are distinct from other neuroectodermal tumours, such as neuroblastoma or Ewing’s sarcoma, demonstrating that this lesion is a distinct entity.
Microscopy
Melanotic neuroectodermal tumour consists of slit-like or larger spaces lined by cuboidal, melanin-containing cells in a fibrous stroma. Small round cells, which are non-pigmented, form clusters in the stroma or lie in the alveolar spaces (Figs 9.13 and 9.14). Some degree of atypia and infrequent mitotic figures are often seen. The round cells resemble neuroblasts and may be associated with neurofibrillary material indicating neural differentiation.
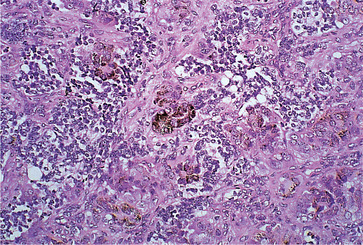 |
| Fig. 9.13
Melanotic neuroectodermal tumour of infancy. Pale pink-staining epithelial cells, some of which are pigmented (centrally), arranged in clusters and surrounded by small, round, darkly staining tumour cells.
|
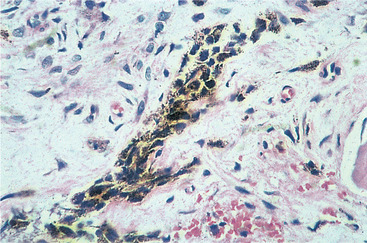 |
| Fig. 9.14
Melanotic neuroectodermal tumour of infancy. Higher power showing the melanin pigment granules within a strand of epithelium.
|
Ultrastructurally, pigmented cells show both epithelial and melanocytic features. Cells with melanosomes in various stages of maturation, and cells with neuroblastic features, including neurosecretory granules and cytoplasmic processes may be seen.
Frequent mitotic figures and loss of differentiation of the melanocyte-like cells suggest more aggressive behaviour. In a tumour that recurred and later disseminated widely after treatment, only the melanocyte-like cells remained at autopsy.
Management
Radiography, CT scans and MRI are not diagnostic in themselves, but the appearance of a destructive maxillary tumour in the maxilla in an infant, together with submucosal pigmentation and high levels of urinary vanillylmandelic acid are distinctive. Serum levels of epinephrine, norepinephrine, vanillylmandelic acid and neuron-specific enolase may be high but typically return to normal after treatment. The clinical and radiographic features of melanotic neuroectodermal tumour of infancy are so distinctive that preliminary biopsy which may provoke the tumour into greater activity should not be necessary.
These tumours are frequently described as benign, but up to 15% may recur and a very few have metastasised and disseminated widely. Histology is an unreliable guide to behaviour, as even those tumours which show histologically malignant features may do well. Nevertheless, a high mitotic index, and possibly CD99 expression, may indicate more aggressive behaviour.
Surgery is the treatment of choice, but the extent of the resection is controversial and can range from local excision and curettage to total maxillectomy. The tumour is non-encapsulated but usually separates easily from the bone at operation. Conservative treatment is usually curative but, even when excision is incomplete, recurrence is rare. Regional lymph node dissection may be necessary only for rare cases where there has been lymphatic invasion. Radiotherapy and chemotherapy (undesirable in an infant) have been shown to be ineffective in controlling this tumour.
Key features are summarised in Box 9.4.
Box 9.4
Melanotic neuroectodermal tumour of infancy: key features
• Very rare
• Congenital or appears in first few months
• Presents as expansion of anterior maxilla, typically bluish in colour
• Benign but destroys bone
• Usually responds to conservative excision or curettage
MALIGNANT NEOPLASMS OF BONE
Osteosarcoma → Summary p. 190
Osteosarcoma is highly malignant and the most common primary (non-odontogenic) neoplasm of bone, but overall is rare, especially in the jaws.
Osteosarcomas rarely have identifiable causes but a few develop late in life after irradiation or Paget’s disease of bone, but the latter type rarely affects jaws.
Osteosarcoma of the jaws is typically seen between the ages of 30 and 40. Males are slightly more frequently affected and the body of the mandible is a common site. There is typically a firm swelling which grows noticeably in a few months and becomes painful (Fig. 9.15). Teeth may be loosened and there may be paraesthesia or loss of sensation in the mental nerve area. Metastases to the lungs may develop early.
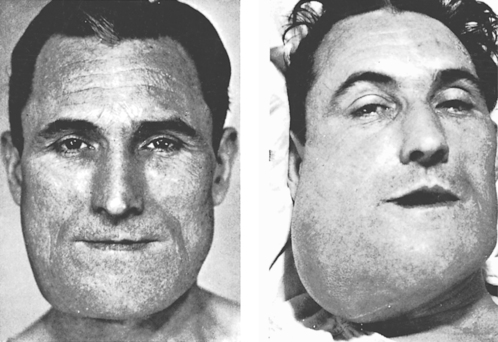 |
| Fig. 9.15
Osteosarcoma. The picture shows the progress of the tumour over a period of 3 months. The patient died with metastases in the lungs, about a month after the second picture was taken. (Taken before the advent of colour photography.)
|
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


