9
Clinical Connections
Inflammation of the tooth pulp (pulpitis)
Response of the pulp to dental caries
“Hey King A . . . the walls have been breached . . . all hands on deck (oops, wrong service) . . . whatever will we do now?” (that’s “soldier speak”). And the great king hollered from the parapet to his troops below:
“Dental caries is a localized, progressive destruction of tooth structure and the most common cause of pulp disease.”
For caries to develop, specific bacteria must become established on the tooth surface. Products of bacterial metabolism, notably organic acids and proteolytic enzymes, cause the destruction of enamel and dentin. Bacterial metabolites diffusing from the lesion to the pulp are capable of eliciting an inflammatory reaction. Eventually, extensive involvement of dentin results in bacterial infection of the pulp.
Injury to odontoblasts
As bacteria penetrate the enamel and enter the dentin, the underlying odontoblasts are adversely affected. Initially, they are stimulated to produce collagen, and there is an accompanying increase in metabolic and enzymatic activity. Soon, however, even before inflammatory changes appear in the pulp, the size and number of odontoblasts is decreased (Fig 9-1). At the same time, their metabolic activity is reduced, and collagen synthesis is depressed. Although they are normally tall columnar cells, odontoblasts affected by caries are transformed into flat or cuboidal cells.
Eventually, the primary odontoblasts die as a result of injury. This usually evokes the formation of a dead tract. A dead tract is an area of dentin that is devoid of odontoblast processes because the odontoblasts have undergone necrosis. Primary odontoblasts that have died are replaced by cells derived from pulpal fibroblasts that migrate from the cell-rich zone of the pulp and differentiate into new odontoblasts. These “replacement” odontoblasts then deposit reparative dentin over the pulpal aspect of the dead tract.
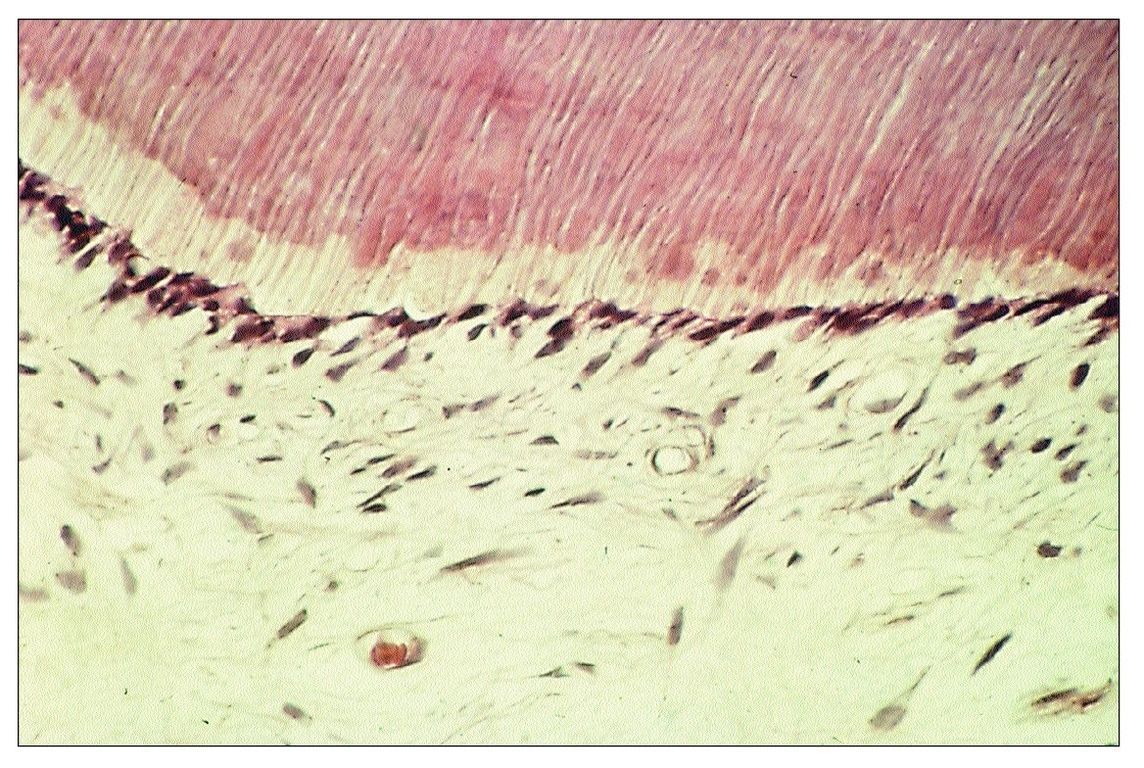
Fig 9-1 Odontoblasts that have been reduced in size as a result of injury associated with caries.
Bacterial antigens diffusing through the dentinal tubules and into the pulp evoke an immune response. Eventually, bacteria proliferate deep into the dentinal tubules and infect the pulp.
Basic reactions that tend to protect the pulp against caries involve:
- A decrease in the permeability of the dentin (dentinal sclerosis)
- The formation of new dentin (reparative dentin)
- Inflammatory and immunologic reactions
Inflammation evoked by caries
Clinically, diagnosis of the extent of pulpal inflammation beneath a carious lesion is difficult. Many factors play a role in determining the nature of the carious process; the individuality of each carious lesion should be recognized. The response of the pulp may vary depending on whether caries progresses rapidly, relatively slowly, or not at all (arrested caries). Moreover, caries tends to be an intermittent process, with periods of rapid activity alternating with periods of quiescence. The rate of carious attack can be influenced by any or all of the following:
- Age of the host (mature enamel is more highly calcified than immature enamel)
- Composition of the tooth, particularly fluoride content
- Nature of the bacterial flora in the lesion
- Rate of salivary flow (patients with xerostomia generally develop rampant caries)
- Antibacterial substances in the saliva (eg, IgA antibodies, lysozyme)
- Oral hygiene (plaque removal)
- Cariogenicity of the diet (refined fermentable carbohydrates) and the frequency with which acidogenic foods are ingested
- Caries-inhibiting factors in the diet (eg, phosphates, calcium-containing foods, chocolate perhaps?)
Dental caries is a protracted process, and lesions progress slowly over a period of years. Consequently, it is not surpris-ing that pulpal inflammation evoked by carious lesions begins insidiously as a low-grade, chronic response rather than an acute inflammatory reaction. The chronic response represents activation of the immune system by bacterial antigens reaching the pulp from the carious lesion.
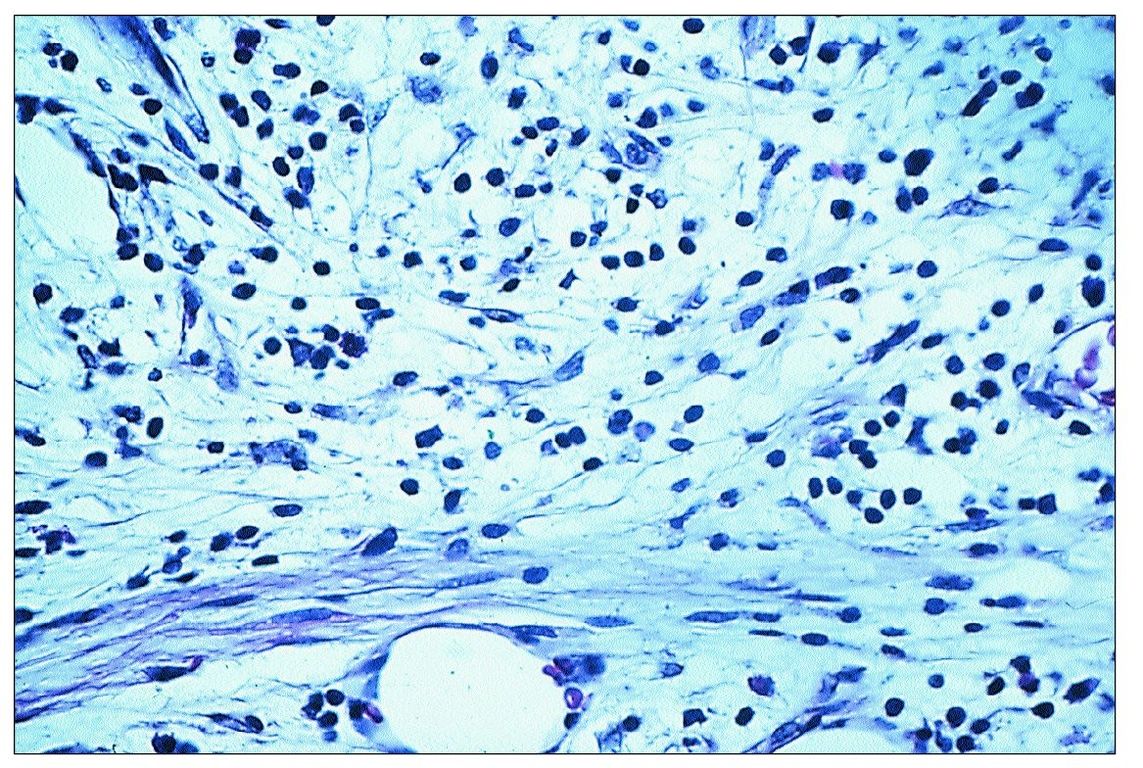
Fig 9-2 Inflammatory cell infiltrate beneath a carious lesion. Note the presence of chronic inflammatory cells and collagen fibers.
Recently, dendritic cells have been identified in the odontoblast layer of the pulp. In a normal pulp, it is thought that these cells play a role in immunosurveillance. Presumably in caries they facilitate immunologic reactions by capturing and processing antigens entering the pulp from the carious lesion. From the pulp, the processed antigen is transported to lymph nodes and presented to T-helper cells. Pulpal macrophages are also able to process and present antigen.
The initial inflammatory cell infiltrate consists almost entirely of lymphocytes, plasma cells, and macrophages (Fig 9-2). Additionally, there is a proliferation of small blood vessels and fibroblasts, and the deposition of collagen fibers. This pattern of inflammation is generally regarded as a cellular immune response.
Remember that chronic inflammation does not necessarily result in permanent damage to tissue; it is similar in many respects to the process of repair. Thus, if the bacteria in the carious lesion are eliminated or inactivated before the pulp becomes infected, connective tissue repair will replace the chronic inflammatory elements. However, in a deep carious lesion, the removal of carious dentin may cause a pulp exposure and further traumatize the pulp. Proper management of a deep carious lesion requires sound clinical judgment and well-developed operative skills.
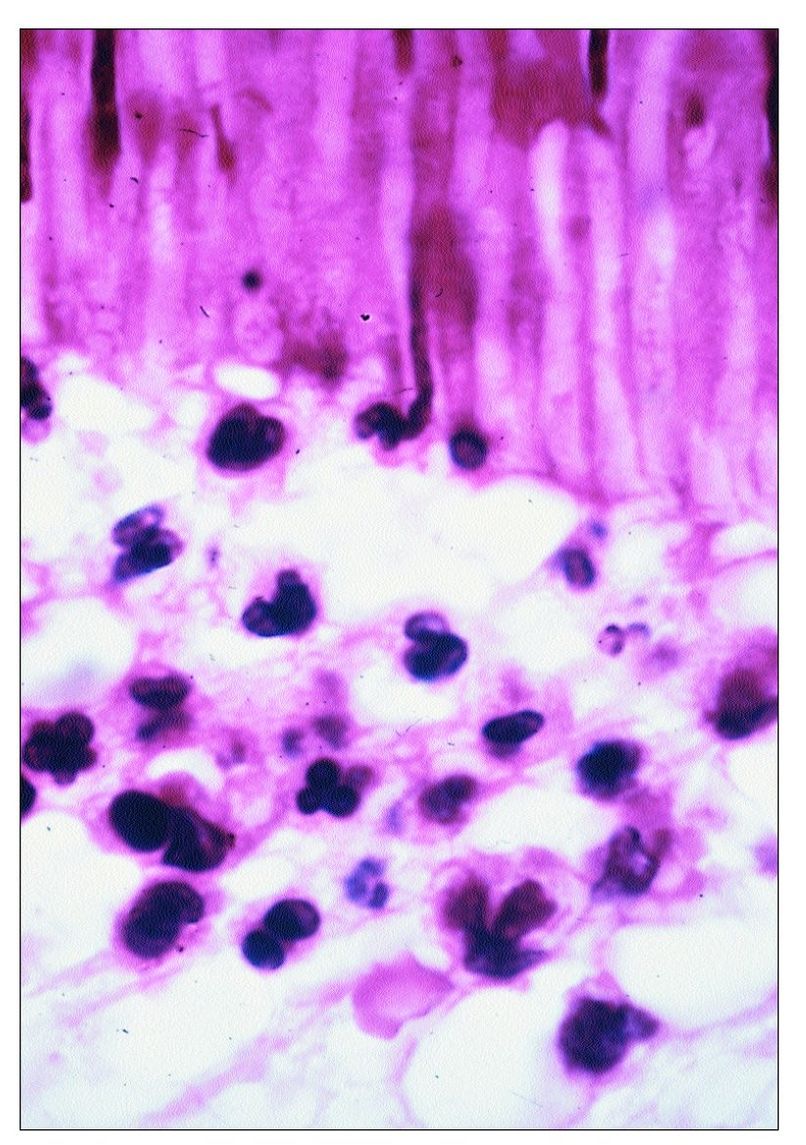
Fig 9-3 Neutrophils accumulating beneath a carious lesion. Some can be seen entering the dentinal tubules.
The severity of pulpal inflammation beneath a carious lesion depends to a great extent on two factors: (1) the depth of bacterial penetration, and (2) the degree to which dentin permeability has been reduced by dentinal sclerosis and/or reparative dentin formation. Usually, if the distance between the invading bacteria and the pulp (including the thickness of reparative dentin) is greater than 1 mm, the inflammatory response is mild. However, by the time the lesion is within 0.5 mm of the pulp, chronic inflammation generally progresses to acute inflammation. This invariably occurs once the reparative dentin beneath the lesion is invaded by bacteria. By this time, the layer of replacement odontoblasts has been destroyed and replaced by inflammatory cells (Fig 9-3).
With the onset of acute inflammation there is vasodilatation, increased vascular permeability, and the accumulation of neutrophils. Neutrophils migrate from blood vessels in response to chemotactic factors derived from the invading bacteria and complement activation. So avid is the neutrophil response that many neutrophils migrate up into the dentinal tubules (see Fig 9-3).
Chronic hyperplastic pulpitis (pulp polyp)
This condition occurs most often in primary and immature permanent teeth with incompletely formed roots. At this stage of development, numerous blood vessels supply the pulp through the wide apical foramen. Because of its rich blood supply and high degree of cellularity, the young pulp is better able to resist bacterial infection than an older pulp. When caries reaches the pulp and produces a pulp exposure, an acute inflammatory reaction ensues. As the pulp exposure increases in size, it may eventually produce a large open cavity. This opening provides a pathway for drainage of the inflammatory exudate. Once drainage is established, acute inflammation subsides and chronic inflammatory tissue proliferates up through the opening created by the exposure. This proliferation results in the formation of a pulp polyp (Fig 9-4). Eventually the polyp becomes epithelialized as desquamated epithelial cells from the oral mucosa are “grafted” onto the surface of the proliferating connective tissue.
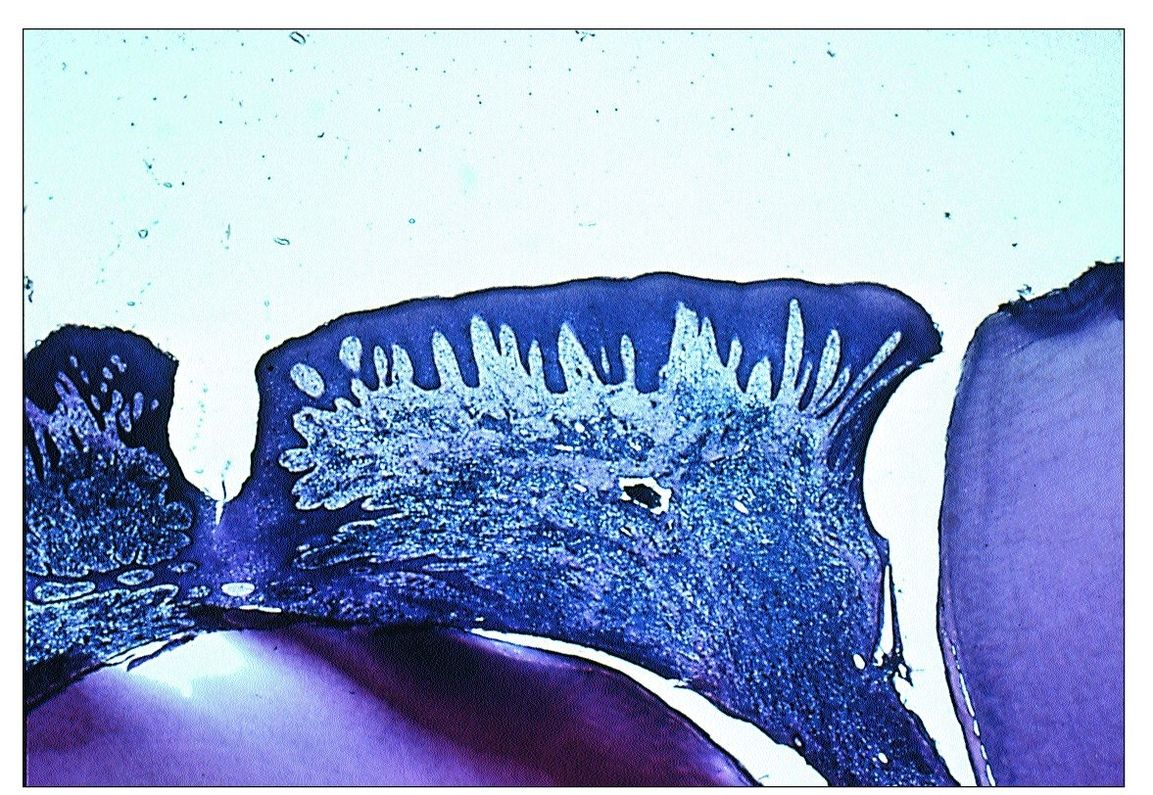
Fig 9-4 Pulp polyp.
Repair in the pulp
The inherent healing potential of the pulp is well recognized. As in all other connective tissues, repair of tissue injury commences with debridement by macrophages followed by proliferation of fibroblasts and capillary buds and by the production of collagen. Because of the open apex of a young tooth, the apical pulp is larger than that of a mature tooth. This allows more blood vessels to enter through the apical foramen. The highly cellular and vascular pulp of a young tooth should have a better healing potential than the pulp of an older tooth, but proof of this is lacking.
In pulpal injury the presence of bacteria severely hampers repair. In experiments on germ-free rats, mechanical exposure of the pulp was followed by complete healing and deposition of reparative dentin beneath the exposure site, whereas the same procedure in conventional animals (ie, not germ-free) generally resulted in total pulp necrosis.
Reparative dentin (RD)
Developmental (primary) dentin is formed during tooth development. When the tooth comes into functional occlusion, the rate of dentin formation is greatly reduced in the coronal portion of the tooth (the root is still forming). Dentin that forms following the completion of tooth formation is termed sec – ondary dentin. Secondary dentin continues to form at a slow rate throughout the life of the tooth, gradually reducing the size of the pulp chamber.
Reparative dentin differs from developmental and secondary dentin because it is produced only in response to specific stimuli. In the case of caries, it forms on the dentin wall beneath the carious lesion. Irritants include extensive wear of the tooth surface, erosion, cracks in the enamel and dentin, dental caries, loss of cementum from the root surface, and dental operative procedures. Thus, RD represents a defense mechanism against loss of enamel, dentin, or cementum.
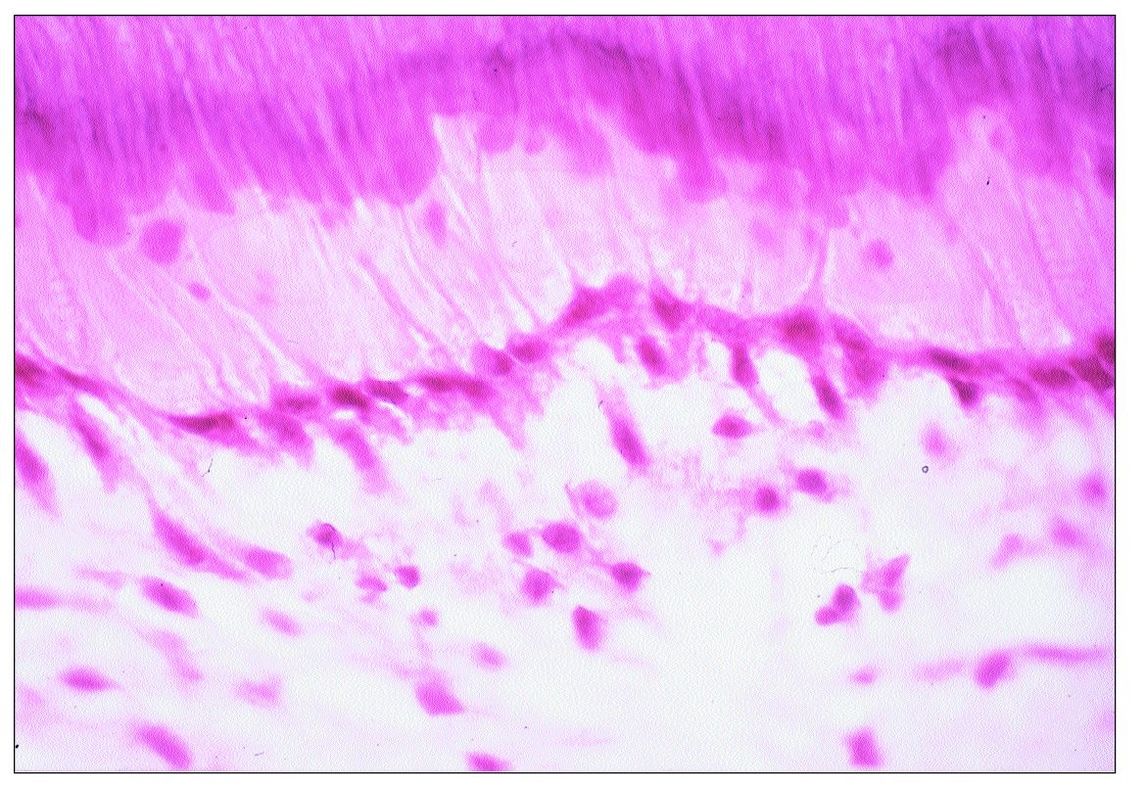
Fig 9-5 Replacement odontoblasts.
RD is formed by cells that replace primary odontoblasts that have been destroyed. Morphologically, these cells are flat to cuboidal in shape, and the odontoblast layer they form has a lower density of cells than the original odontoblast layer (Fig 9-5).
Following the loss of primary odontoblasts, there is a time lag of about 20–40 days before RD formation commences. During this time the following events are taking place: (1) odontoprogenitor cells in the underlying pulp undergo mitosis; (2) the daughter cells migrate to the site where RD will be formed; (3) they differentiate into preodontoblasts and commence to secrete dentin matrix; and (4) the preodontoblasts further differentiate into odontoblasts.
As compared with primary dentin, RD is generally less tubular and less well calcified. In some cases no tubules are formed. The quality of RD (ie, the degree to which it resembles primary dentin) is highly variable. Factors influencing its formation include the nature and magnitude of the stimulus (injury or irritant) and the status of the pulp. If the pulp is healthy, the RD is of good quality. If the pulp is inflamed, the quality is poor. At times, RD is laid down so haphazardly that areas of soft tissue become entrapped within the developing matrix, thus producing a “Swiss cheese” pattern.
Is RD protective, or is it simply a form of scar tissue? Available evidence indicates that it actually does shield the pulp from injury. In the case of caries, RD often helps the pulp survive. It seems that operative procedures are better tolerated in teeth having a layer of preformed RD beneath the cavity preparation than in teeth without RD. However, it must be emphasized that poor-quality RD may not shield the pulp from injury.
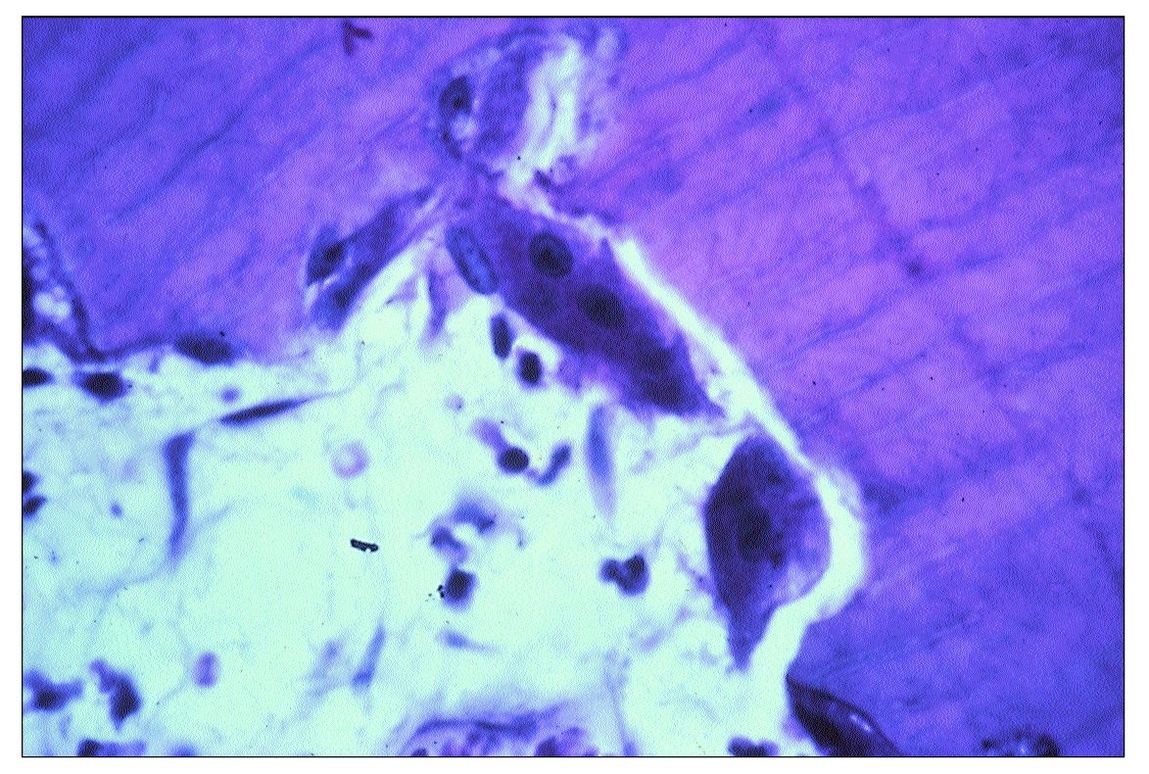
Fig 9-6 Two osteoclasts in a resorption bay in dentin.
Why can RD be protective? As pointed out earlier, there is often an atubular zone at the interface between primary and reparative dentin that presumably limits the diffusion of irritants through the dentin, thus blocking the inward diffusion of irritants.
Internal resorption of dentin
Two types of resorption affect teeth: internal and external. The former involves the pulp chamber and root canals, whereas the latter affects the external surface of the root. The etiology of internal resorption remains speculative. A few cases have been reported where internal resorption occurred in healthy teeth, even in unerupted teeth. Most often, resorption occurs in chronically inflamed pulps where lymphocytes are present and there is proliferation of small blood vessels. Resorption bays containing osteoclasts can be observed on the surface of the dentin (Fig 9-6).
Internal resorption appears to be an intermittent process with periods of active resorption alternating with periods of inactivity. During periods of remission, mineralized tissue is often deposited within the areas of resorption.
Which inflammatory stimuli initiate internal resorption? There are at least three possibilities: (1) cytokines such as IL-1 and TNF, (2) prostaglandin E2, and (3) increased tissue pressure.
Internal resorption is usually detected radiographically, although sometimes it manifests as a pinkish hue on the tooth surface (due to the underlying vascular tissue). The resorptive defect is always larger than it appears on radiographs. Once diagnosed, root canal treatment should be instituted, as resorption is usually progressive and the pulp must be removed to halt the process. Untreated, resorption may perforate the crown and provide a portal of entry for microorganisms.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


