Eponymous conditions
Abrikossof tumour Granular cell myoblastoma (tumour).
Albers-Schonberg disease Osteopetrosis (p. 405).
Alstrom syndrome Congenital nerve deafness and retinitis pigmentosa.
Avellis syndrome A unilateral paralysis of the larynx and palate.
Battle sign Bruising over the mastoid bone – sign of a basilar skull fracture.
Beckwith–Wiedemann syndrome Congenital gigantism, and omphalocoele or umbilical hernia.
Beeson sign Myalgia, facial oedema and fever in trichinosis.
Behçet syndrome ‘Adamantiades syndrome’ (see Ch. 36).
Bell palsy The common lower motor neurone facial palsy (see Ch. 37).
Bell sign Seen in lower motor neurone facial palsy, when the eye rolls upward on attempted closure.
Biemond syndrome Congenital obesity and hypogonadism.
Binder syndrome Congenital maxillonasal dysplasia, and absent or hypoplastic frontal sinuses.
Blackfan–Diamond syndrome Congenital red cell aplasia.
Bloom syndrome Congenital telangiectasia, depigmentation and short stature.
Cannon disease Congenital white sponge naevus.
Carabelli cusp Congenital additional palatal cusp on upper molars.
Christmas disease Blood clotting factor IX defect.
Clutton joints Symmetrical hydrarthrosis of knees in congential syphilis, appearing around puberty.
Cockayne syndrome Premature ageing, dwarfism, deafness and neuropathy.
Coffin–Lawry syndrome Congenital osteocartilaginous anomalies and learning disability.
Cross syndrome Athetosis, learning disability, gingival fibromatosis and hypopigmentation.
Destombes–Rosai–Dorfman syndrome Rosai–Dorfman syndrome.
Duhring disease Dermatitis herpetiformis.
ECHO viruses Enteric cytopathogenic human orphan viruses.
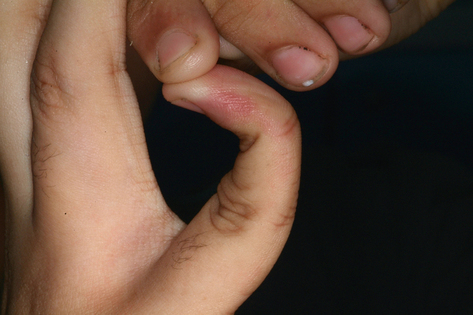
Ehlers–Danlos syndrome A group of congenital collagen disorders (autosomal dominant, autosomal recessive, or X-linked), with altered mechanical properties of skin, joints, ligaments and blood vessels. Phenotypes vary depending upon which collagen type is affected. EDS is characterized by hyperflexible joints, hyperextensible skin, bleeding and bruising, and mitral incompetence. Patients can bend the thumb right back (Fig. 56.1) and may be able to touch the tip of their nose with their tongue. Recurrent dislocation of the temporomandibular joint may be seen. Dental anomalies include deep-fissured premolars and molars, dentinal abnormalities, such as shortened deformed roots, and multiple large pulp stones. Ten types were described: in types IV, VIII and IX there is severe early onset periodontal disease with loss of permanent teeth. Type III genotypes show resistance to local analgesia.
Fanconi anaemia Congenital anaemia, abnormal radii and risk of oral carcinoma and leukaemia.
Felty syndrome Rheumatoid arthritis and neutropenia.
Filatov disease Infectious mononucleosis.
Fitzgerald–Gardner syndrome Gardner syndrome
Fordyce disease (Fordyce spots) See Ch. 24.
Froehlich syndrome Congenital obesity, hypogonadism, and risk of learning disability and open bite.
Garré osteomyelitis This is proliferative periostitis (p. 405).
Gasserian ganglion The trigeminal ganglion.
Gilles de la Tourette syndrome Coprolalia (utterance of obscenities).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


