5
Examination of the Oral Cavity
Dentists should have a special interest in the physical examination of the oral cavity since the mouth is the anatomical area of the body for which they are the ultimate authority. Therefore, the organization of this section is more detailed and provides greater emphasis on possible findings and interpretation of data. Basic instrumentation for the oral examination includes a good light source, a mouth mirror, an explorer, a periodontal probe, dry gauze sponges, and an air syringe. The need for specialized instrumentation and additional diagnostic procedures will vary with the findings and differential diagnoses developed.
Examine the Vermilion of the Lips
The mouth begins at the mucocutaneous junction of the vermilion border of the lips. The vermilion border, a zone of specialized non-mucus-producing tissue, is bounded by the facial skin and the moist labial mucosa of the mouth. With age and exposure to the elements, the color of the vermilion border may change from a pink or red (vermilion) to a bluish hue. This region is a common site for physiological pigmentation, ephelides or freckles, and rarely, pigmentation suggestive of Peutz-Jeghers syndrome. Varices, hemangiomas, and rarely, telangiectasias (hereditary hemorrhagic telangiectasia and CREST syndrome) may be noted.
Swelling of the lip may indicate cellulitis of dental origin or angioedema. Vesicular lesions of the lip may represent the initial phase of recurrent herpes labialis (RHL) (see “Herpetic Infections”). Serohemorrhagic crusting of the lips is highly suggestive of erythema multiforme, an acute vesiculoulcerative disorder. Thickening of the vermilion border with the development of vertical fissures and/or the loss of distinction of the mucocutaneous junction may be indicative of actinic cheilosis. The upper and lower lips join at the commissures. Crusting or weeping erosion in this area is characteristic of angular cheilitis.
Peutz-Jeghers Syndrome
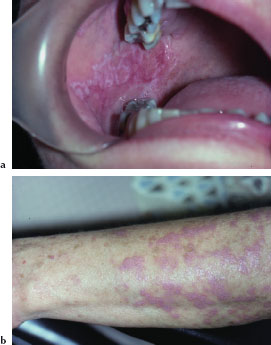
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disorder characterized by mucocutaneous melanin pigmentation and unique hamartomatous polyps (Peutz-Jeghers polyps) affecting the gastrointestinal tract. These polyps have a predilection for the small intestine. Mutations of the LKB1 tumor suppressor gene have been demonstrated in up to 80% of cases. PJS patients are at an increased risk of cancer, especially cancer of the colon, stomach, small intestine, pancreas, breast, and ovary. The incidence of PJS is estimated to be 1 in 50,000 to 1 in 200,000.
Clinical Features
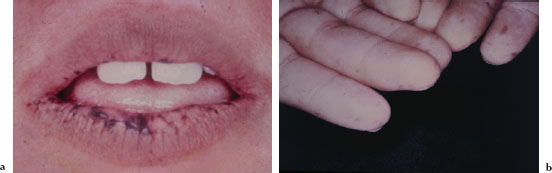
The characteristic mucocutaneous pigmentation presents as 1–5 mm dark brown or blue macule affecting the lip vermilion (Figure 5.1a), buccal mucosa, hands (Figure 5.1b), and feet. These macules typically develop in infancy, but may fade with age. Since these areas of pigmentation are asymptomatic, their presence is often underappreciated. Underlying gastrointestinal symptoms, such as intestinal obstruction, abdominal pain, and bloody stools, often prompt the patient to seek medical attention.
Diagnosis
The diagnosis of PJS is straightforward and established by the presence of either two or more Peutz-Jeghers polyps, one Peutz-Jeghers polyp and mucocutaneous pigmentations, or one Peutz-Jeghers polyp and a positive family history for PJS.
Figures 5.1a and b. Peutz-Jeghers syndrome.
Treatment
There is no cure for PJS. The increased cancer risk associated with PJS mandates earlier and more frequent screening for malignant disease. The mucocutaneous pigmentation requires no treatment.
Hereditary Hemorrhagic Telangiectasia
Hereditary hemorrhagic telangiectasia (HHT) is an uncommon autosomal dominant fibrovascular disorder. Also known as Rendu-Osler-Weber disease, HHT is characterized by the triad of mucocutaneous telangiectasias, arteriovenous malformations, and recurrent hemorrhagic episodes. The morbidity and mortality of HHT is mainly attributable to the arteriovenous malformations (AVMs), which may occur in the lungs, skin, brain, liver, and gastrointestinal tract. Mutations underlying HHT have been identified in two different genes: edoglin (ENG) and activin-receptor-like-kinase (ALK1). For either case, homozygous forms of HHT are considered lethal. The incidence of HHT is estimated to be 1 in 5,000 to 1 in 8,000.
Clinical Features
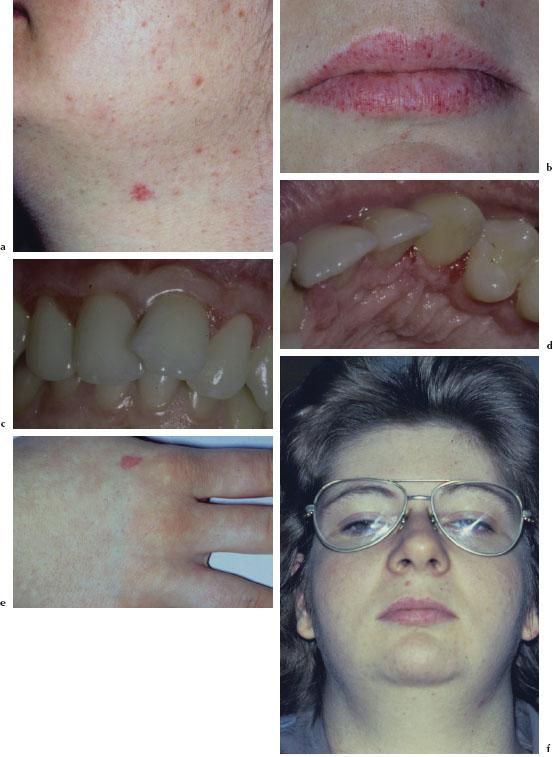
The most common presenting feature of HHT is recurrent epistaxis, which usually occurs between the ages of 10 and 20 years and may progress in frequency and severity with age. The telangiectasias become apparent in the third to fourth decade of life and usually progress both in size and number with age. They present as small flat or slightly raised bright red-purple lesions. These lesions blanch upon pressure and may be found on the face (Figure 5.2a), conjunctivae, lips (Figure 5.2b), tongue, palate, gingivae (Figure 5.2c and 5.2d), fingers (Figure 5.2e), arms, and trunk. Gastrointestinal involvement may result in severe blood loss leading to severe anemia with pallor (Figure 5.2f). AVMs are associated with pulmonary shunting, transient ischemic attacks, brain abscess, and hepatic shunting.
Diagnosis
The diagnosis is based on the presence of three of the following four findings: spontaneous recurrent epistaxis, mucocutaneous telangiectasia, visceral involvement, and an affected first-degree relative.
Treatment
There is no cure for HHT, and current treatment strategies entail close monitoring to identify and treat a potentially lethal AVM. Symptomatic treatment of epistaxis is often required, as is iron supplementation to manage anemia.
Figures 5.2a–f. HHT.
Angioedema
Angioedema (AE) is a reversible localized swelling of the deep subcutaneous or submucosal tissues secondary to increased vascular permeability caused by locally produced vasoactive substances such as histamine, tryptase, prostaglandin F2α, and bradykinin. While there are many established causes for AE (Table 5.1), the cause in most cases remains unknown. The true incidence of AE is unknown, but it is postulated that up to 20% of the population will experience at least one episode of AE in a lifetime.
Table 5.1. Causes of angioedema.
| Mechanism of disease | Precipitating factors |
| IgE mediated allergic response | Insect venom, shellfish, peanuts, antibiotic, NSAIDs |
| Pseuodoallergic response | Opioids, polymyxin, ACE inhibitors |
| Autoimmune disease | Hashimoto thyroiditis |
| C1 esterase inhibitor (C1-INH) deficiency | Hereditary, acquired, idiopathic |
| Trauma | Mechanical, chemical, thermal |
Clinical Features
While any part of the body may be affected, most cases are characterized by edema of the face and/or pharyngeal tissues (Figures 5.3a and 5.3b). The affected skin or mucosa may be tender and warm and the patient may complain of a burning sensation. Severe AE affecting the head and neck may spread to the larynx, resulting in life-threatening airway obstruction. About 50% of patients also manifest urticaria and pruritis. Involvement of the abdominal viscera is often associated with severe pain.
Diagnosis
The abrupt onset of AE affecting an observable area is easily recognized and diagnosed.
Treatment
The initial treatment often focuses on providing symptomatic relief. AE presenting with urticaria usually responds well to antihistamines and corticosteroids. Efforts to identify the cause of the AE should be undertaken, and patients with recurrent AE should be referred to an allergist or dermatologist for a comprehensive evaluation.
Figures 5.3a and b. Angioedema.
Erythema Multiforme
Erythema multiforme (EM) is an acute, typically self-limiting, and potentially recurrent mucocutaneous vesiculoerosive disorder. Severity varies from mild (EM minor) to moderate (EM major) to potentially fatal (Stevens-Johnson syndrome [SJS] and toxic epidermal necrolysis [TEN]) (Table 5.2). Some authorities consider oral EM minor/ EM major and SJS/TEN to be two distinct disease processes.
The etiology of EM is most likely due to a genetically predisposed allergic host response to antigenic challenge (Table 5.3). Most cases of EM minor and EM major are related to an infectious agent, typically herpes simplex virus, while most cases of SJS and TEN are related to a pharmacologic agent, most frequently a sulfonamide, an anticonvulsive drug, or a COX-1 inhibitor. In many cases the causative agent is not identified. The incidence of EM minor and EM major is unknown, while the incidence of SJS and TEN is estimated to be two to three cases per million.
Clinical Features
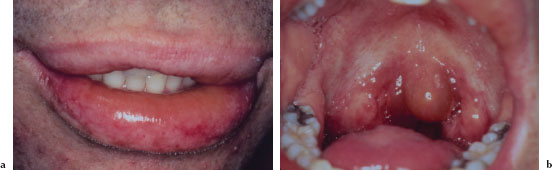
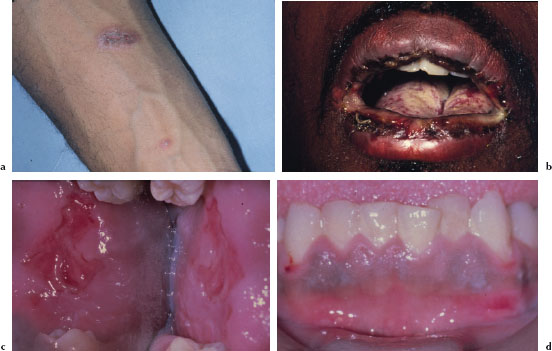
Cutaneous lesions usually begin as erythematous papules that progress to form the more characteristic iris or target lesions. These lesions typically arise in an acral distribution (Figure 5.4a). In more severe forms of EM, the cutaneous lesions may present as more widespread erythematous or purpuric macules and blisters. Such patients may demonstrate a positive Nikolsky’s sign. Hemorrhagic crusting of the lips is highly characteristic and virtually pathognomonic (Figure 5.4b). Intraorally, lesions on the unattached mucosal tissues predominate (Figure 5.4c and 5.4d). In the vast majority of cases, mucosal lesions tend to appear abruptly and manifest as painful vesicles, ulcerations, and erosions. Mucocutaneous lesions tend to heal completely in 2–6 weeks.
Table 5.2. Spectrum of erythema multiforme.
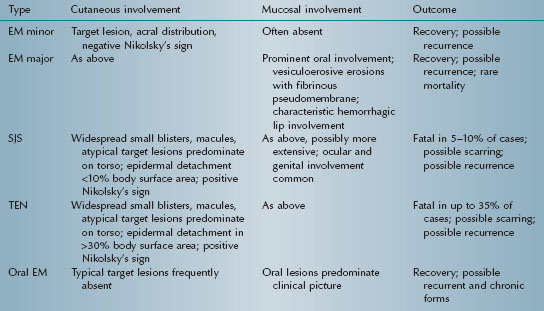
Table 5.3. Some causes of erythema multiforme.
| Infectious agents | Drugs |
| β-Hemolytic streptococci | Barbiturates |
| Coccidiomycosis | Carbamazepine |
| Coxsackie virus | Cephalosporins |
| Diptheria | Clotrimazole |
| Epstein-Barr virus | Fluoquinolones |
| Herpes simplex 1 & 2 | NSAIDs |
| Herpes zoster | Penicillin |
| Influenza, type A | Phenytoin |
| Mumps | |
| Mycoplasma pneumoniae | |
| Vaccinia |
Figures 5.4a–d. Erythema multiforme.
Diagnosis
The typical abrupt onset, combined with the presence of the characteristic mucocutaneous lesions (i.e., target lesions, crusted lips, and vesiculoulcerative oral lesions) is diagnostic. Historical evidence of prior occurrence and/or exposure to a possible causative drug or infectious agent reinforces the diagnosis. For equivocal cases, a biopsy and immunofluoresence studies may be useful to rule out other conditions in the differential diagnosis. Conditions that may mimic EM include erosive lichen planus, pemphigus, mucous membrane pemphigoid, lupus erythematosus, herpetic gingivostomatitis, major aphthous, and hand-foot-and-mouth disease.
Treatment
Most cases of EM resolve in 2–6 weeks, and treatment is generally palliative and supportive. Efforts to ensure adequate hydration and nutrition are mandatory, as is close monitoring. Anesthetic mouth rinses such as diphenhydramine hydrochloride and viscous lidocaine may be prescribed for oral pain, along with a bland soft nutritious diet. Antibiotic therapy may be indicated if secondary infection becomes evident. The withdrawal of any suspected causative medication should be undertaken and a careful history should be obtained to identify other possible underlying causes. Suspected cases of SJS or TEN mandate immediate referral to a physician.
Actinic Cheilosis
Actinic cheilosis is essentially actinic keratosis affecting the lip vermilion. Etiologic factors include cumulative ultraviolet radiation (UVR) exposure, skin phenotype, age, male sex, outdoor occupation, tobacco habits, and host immunological status. It represents the early clinical stage of a continuum that ultimately may progress to squamous cell carcinoma of the lip. Cumulative exposure to UVR in sunlight is the most important cause of actinic cheilosis. The true incidence of actinic cheilosis is unknown.
Clinical Features
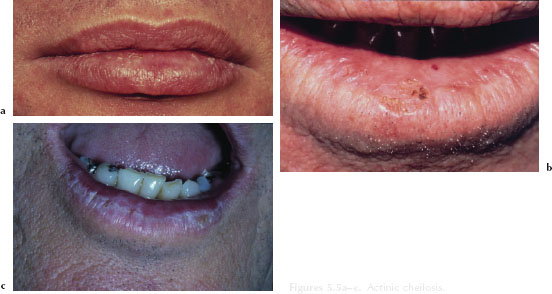
Actinic cheilosis typically develops over several years and primarily affects the lower lip. Initially, the patient may manifest a dry, unobtrusive chapped lip (Figure 5.5a). More advanced cases manifest marked parallel folds, isolated hyperkeratotic plaques, and a loss of the definition of the vermilion-cutaneous border (Figure 5.5b). In later stages, actinic cheilosis may appear mottled or opalescent in color, with a slightly elevated white or gray plaque (Figure 5.5c). The waxing and waning of erythematous or hemorrhagic areas over a prolonged period of time may represent an ominous sign.
Diagnosis
The working diagnosis of actinic cheilosis usually is straightforward and is derived by correlating the history with clinical findings in an at-risk patient. The presence of concurrent actinic keratoses on sun-exposed areas (i.e., face, neck, bald scalp, and ears) reinforces the diagnosis. Lesions that persist or do not respond to preventive measures should be biopsied. Unfortunately, the clinical appearance of actinic cheilosis does not always correlate directly with the underlying histological changes. Suspicious-looking lesions may prove to be remarkably benign, while a small area of actinic cheilosis may in fact represent severe dysplasia or even squamous cell carcinoma.
Figures 5.5a–c. Actinic cheilosis.
Treatment
Commonly attempted ablative therapies to remove actinic cheiloses include cryotherapy with liquid nitrogen, topical 5-fluorouracil (5-FU), excision, laser ablation, and chemical peel. Given the strong etiologic link between UVR and actinic cheilosis, reducing exposure to sunlight (or other forms of UVR) is the single most important measure for preventing actinic cheilosis. Avoiding peak sun exposure; covering up exposed skin; wearing a hat that shades the neck, face, and ears; wearing sunglasses; and using a sunscreen with a sun protection factor (SPF) of 15 or higher is recommended for all patients.
Angular Cheilitis
Angular cheilitis (perlèche) is an erythematous fissuring of the commissures of the mouth and adjacent skin. The lesions are typically infected with Candida albicans and staphylococci or streptococci. Factors that predispose to the development of angular cheilitis include insufficient vertical dimension of occlusion, nutritional deficiencies, endocrinopathies, medications, poor oral hygiene, dental prostheses, and conditions of altered immunity. While angular cheilitis may be an isolated occurrence, it is often associated with intraoral candidiasis. The true incidence of angular cheilitis is unknown, but the number of patients who harbor Candida albicans as a commensal organism in the oral cavity varies from 25% to 75%.
Clinical Features
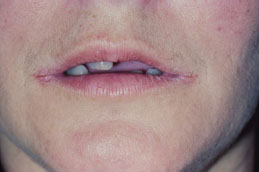
Angular cheilitis presents as unilateral (Figure 5.6) or bilateral (Figure 5.7) erythematous fissures of the commissures of the lips and adjacent skin. The lesion often has a glistening or moist appearance, but variable crusting may also occur.
Figure 5.6. Angular cheilitis.
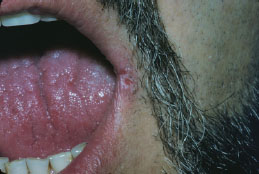
Figure 5.7. Angular cheilitis.
Diagnosis
The characteristic site-specific presentation of angular cheilitis makes for a straightforward diagnosis.
Treatment
Mild cases of angular cheilitis are often transient and resolve uneventfully. More persistent cases typically respond to the use of a topical antifungal/steroid formulation and proper hygiene. For all cases, it is prudent to identify and, when possible, correct any predisposing factors.
Examine the Labial and Buccal Mucosa
To expose the labial mucosa, retract the upper and lower lips. Numerous minor salivary glands are located in this area. Using bidigital palpation, these glands are often apparent as small nodules. Minor salivary glands express saliva through pinpoint ducts. The function of these glands may be evaluated by everting the lips and drying the labial surfaces. Within several seconds, small beads of saliva should be expressed from these ducts. Mucoceles, extravasations of mucous-type saliva (mucin) into the connective tissue, occur frequently in the lower lip.
Still using the fingers as retractors, extend the examination to the buccal and vestibular mucosa. Observe color and other tissue characteristics. Find the parotid duct (Stensen’s duct) located on the buccal mucosa adjacent to the maxillary first and second molar teeth. Assess parotid function. Firmly massaging the gland toward Stensen’s duct should yield a clear fluid. Expression of casts, pus, or blood may indicate the presence of an infection, a sialolith, or a malignancy. Reduced salivary flow, dry mouth or xerostomia, is most often noted secondary to the use of anticholinergic agents, or in association with diabetes mellitus, Sjögren’s syndrome, or head-and-neck radiotherapy.
A common and totally benign finding, linea alba typically presents bilaterally as a grayish-white keratotic linear plaque on the buccal mucosa that runs parallel to the adjacent occlusal plane. It is thought to represent mild occlusal trauma to the buccal mucosa characterized by hyperorthokeratosis (hyperkeratosis without retention of nuclei). More pronounced lesions may exhibit a shredded or shaggy appearance. Such pronounced cases likely indicate that a factitial habit (e.g., habitual cheek chewing) has contributed to the development of the lesion. Treatment is neither required nor recommended for linea alba.
In leukoedema, a normal mucosal variation, the mucosa appears wrinkled and opalescent. Traumatic lesions and recurrent aphthous stomatitis are commonly observed on the labial and buccal mucosa. The buccal mucosa is also a common site for the lacy linear Wickham striae of l ichen planus and the diffuse thickened white lesions of white sponge nevus, a congenital condition. Snuff keratosis is most commonly observed in the mucobuccal fold.
Pigmented lesions affecting the oral tissues are rare. Fordyce granules (ectopic sebaceous glands) appear as clusters of small yellow nodules and are frequently observed in the labial and buccal mucosa. An amalgam tattoo and a hematoma may be observed as an isolated bluish or grayish pigmentation affecting almost any area of the mouth. Examination of the buccal mucosa may also reveal commonly occurring raised or nodular lesions such as a fibroma and a papilloma, or less commonly occurring lesions such as a neuroma, hemangioma, or malignant neoplasm. Other conditions that may affect the labial and buccal mucosa include erythema multiforme and pemphigus vulgaris.
Mucocele
A mucocele (mucous retention phenomenon, mucous extravasation phenomenon) represents a collection of salivary mucin entrapped within the connective tissue. It is thought that microtrauma to a minor salivary gland causes ductal disruption followed by mucin extravasation into the surrounding soft tissue. They are quite common, demonstrate no sex predilection, and occur more frequently in children, adolescents, and young adults. Mucoceles are most frequently located in the lower lip (60–70%), but may develop at any location where minor salivary glands are present, including the soft palate, retromolar region, and buccal mucosa. Their occurrence in the maxillary lip is uncommon.
Figures 5.8a and b. Mucocele.
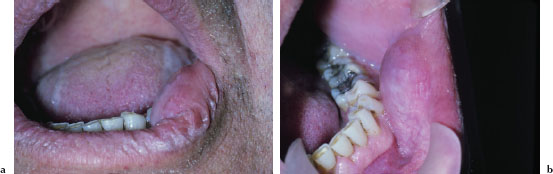
Figure 5.9. Mucocele.
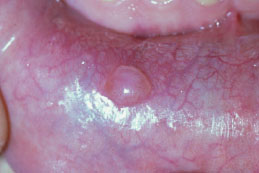
Clinical Features
While some mucoceles may become large enough to cause a visible lip swelling (Figures 5.8a and 5.8b), typically they appear as a discrete, small, translucent, soft, painless swelling of the mucosa (Figure 5.9). Their color may range from normal pink to deep blue. A deep blue color reflects tissue cyanosis as a result of vascular congestion associated with the stretched overlying tissue and the translucent character of the accumulated fluid beneath. A more deep-seated lesion may appear as a normal colored submucosal nodule.
Diagnosis
The diagnosis of a mucocele is based principally on clinical and historical findings. The patient may or may not recall a specific inciting traumatic event. Lesions that may mimic a mucocele include a fibroma, vascular lesion, neural tumor, or salivary gland tumor.
Treatment
Smaller mucoceles often undergo spontaneous involution and resolution. Lesions that persist or interfere with normal function are easily managed surgically. It is important that the surgeon remove all visible minor salivary gland tissue at the time of surgery, in order to reduce the risk of recurrence.
Dry Mouth
Dry mouth is a commonly encountered problem in dental practice. The term used to describe the patient’s subjective complaint of a dry mouth is xerostomia. Numerous etiologic factors such as salivary gland disease, systemic disease, medications, head and neck irradiation, and some chemotherapy regimens contribute to dry mouth. Xerostomia is a common presenting complaint in about 30% of patients over the age of 65 years.
Clinical Features
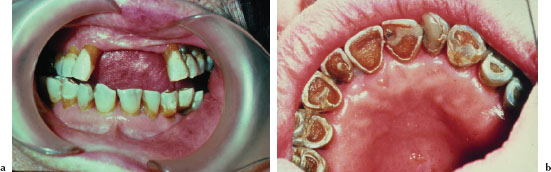
The oral findings associated with dry mouth are variable and depend on the severity andb duration of dryness. Mild acute cases may manifest no overt clinical changes. Common findings of oral dryness include a lack of saliva pooling in the floor of the mouth; inadequate wetting of the mucosal tissues (Figure 5.10a); a dry, furrowed, or fissured tongue (Figure 5.10b); patchy areas of erythema, usually indicative of candidiasis; and rampant caries (Figures 5.11a and 5.11b).
Diagnosis
The diagnosis of a dry mouth is relatively straightforward; the challenge is to identify the underlying cause or causes. Obtaining an exhaustive medical history is mandatory, and further medical evaluation may be necessary.
Treatment
Treatment strategies for dry mouth are individually tailored and largely contingent upon the severity and duration of the dry mouth. Medication adjustment and/or underlying disease management may be all that is necessary to reestablish normal salivary production. Episodic cases, such as may occur with occasional allergy medication ingestion, may be managed with simple supportive measures such as frequent sipping of water throughout the day. In contrast, the more severe and permanent dry mouth associated with head and neck irradiation is often only minimally responsive to currently available management protocols. These protocols consist of improved oral hygiene, saliva substitutes, fluoride therapy, and sialogogue administration.
Figures 5.10a and b. Xerostomia.
Figures 5.11a and b. Xerostomia.
Leukoedema
Leukoedema is a common normal variation of the buccal mucosa. The etiology is unknown, but some authorities suggest low-grade irritation contributes to an increased thickness of the epithelium, intracellular edema in the prickle cell layer, and hyperparakeratosis (hyperkeratosis with retention of nuclei). Prevalence rates vary greatly among geographic locations and among different ethnic groups. It may be present at birth, but is most often diagnosed in adolescence. Prevalence rates in blacks vary from 70% to 90%, while rates in whites vary from 10% to 90%.
Clinical Features
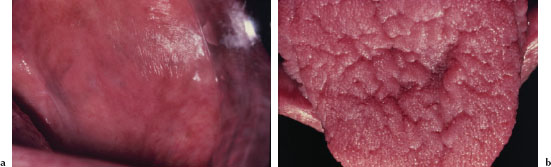
Leukoedema presents as a bilateral white, filmy, or milky opalescence of varying intensity affecting the buccal mucosa (Figure 5.12). The mucosa may appear folded or wrinkled (Figure 5.13). Leukoedema either disappears or undergoes significant diminution when the affected tissue is stretched. Infrequently, leukoedema affects the tongue, lips, and floor of the mouth and similar mucosal changes have been reported affecting vaginal and laryngeal mucosa.
Diagnosis
The tell-tale disappearance upon stretching is diagnostic. Other white lesions that may mimic leukoedema include white spongy nevus, snuff keratosis, lichen planus, proliferative verrucous leukoplakia, and hereditary benign intraepithelial dyskeratosis (Witkop disease).
Treatment
No treatment is necessary for leukoedema. It has no malignant potential and does not change significantly after 25–30 years of
age.
Figure 5.12. Leukoedema.
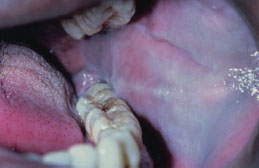
Figure 5.13. Leukoedema.
Recurrent Aphthous Stomatitis
“Aphthous” comes from the Greek word “aphtha,” which means ulcer. Recurrent aphthous stomatitis (RAS) is a recurrent, self-limiting, noninfectious inflammatory disorder of the oral cavity characterized by painful shallow ulcerations. The actual cause of RAS remains unclear, although it is a T-cell-mediated condition and a genetic predisposition is likely. RAS is considered the most prevalent oral mucosal disease, affecting an estimated 20% of the population, and has been associated with numerous other conditions (Table 5.4). “Aphthous stomatitis” has been used interchangeably with “aphthous ulcers” and may be more accurate terminology.
Clinical Features
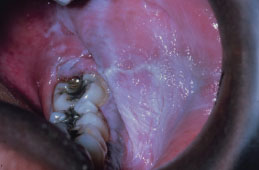
RAS typically presents as one or more well-circumscribed, round or oval, yellowish to white colored, shallow ulcers that are surrounded by an erythematous halo of inflamed mucosa. The lesions are typically painful and may interfere with speaking, eating, and swallowing. Some patients relate a prodromal burning sensation 24–48 hours before lesion onset. RAS characteristically affects the nonkeratinized oral mucosa, such as the labial and buccal mucosa (Figures 5.14, 5.15, and 5.16), tongue (Figure 5.17), and soft palate. RAS may be further characterized according to its clinical appearance and severity (Table 5.5).
Table 5.4. Conditions often associated with RAS.
| Local trauma (factitial or iatrogenic) | Hematological diseases (cyclic neutropenia, leukemia) |
| Stress | Rheumatic disorders (Behget disease, Reiter’s syndrome) |
| HIV infection | Allergy (food, drugs) |
| GI diseases (Crohn’s disease, ulcerative colitis, and celiac disease) | Nutritional deficiencies (iron, folic acid, zinc, and vitamins B1, B2, B6, and B12) |
Figure 5.14. RAS.
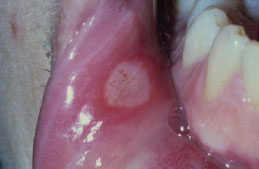
Figure 5.15. RAS.
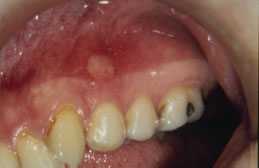
Figure 5.16. RAS.
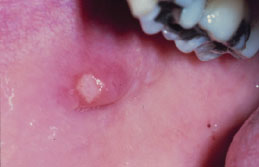
Figure 5.17. RAS.
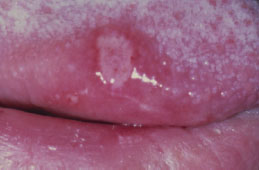
Table 5.5.. Classification of RAS.
| Type | Characteristics |
| Minor RAS | Most common type (80%); single or multiple small ulcers (< 1.0 cm); heal without scarring in 7–10 days. |
| Major RAS | Less common type (10–15%); single or multiple large ulcers (> 1.0 cm); ulcers are deeper and more persistent; heal in several weeks; scarring possible. |
| Herpetiform RAS | Least common (5–10%), multiple small pinpoint ulcerations; clustered or cropped presentation; heal in 1–4 weeks. |
Diagnosis
The diagnosis of RAS is principally established based upon the characteristic clinical presentation and history. Conditions to consider in the differential diagnosis include recurrent intraoral herpes simplex infections, primary herpetic gingivostomatitis, herpes zoster, a traumatic ulcer, EM, and ulcers associated with systemic disease.
Treatment
ntThe goals of therapy are to relieve pain, promote healing, and decrease the frequency and severity of recurrence. Although a variety of treatment modalities (topical and systemic) have been promoted to either eliminate or reduce the duration of recurrence of RAS, their clinical value remains unproven and can be controversial. On the basis of efficacy, cost, and safety, topical steroids remain the treatment of choice for patients with minor RAS. Finally, a comprehensive medical evaluation may be warranted to identify underlying systemic conditions potentially associated with RAS.
Lichen Planus
Lichen planus (LP) is a cell-mediated chronic inflammatory disease involving mucous membranes and skin. It is thought to arise as a result of an immune response to certain unknown antigens within the basal cell layer of the epithelium. LP primarily occurs in middle-aged individuals, although all ages are at risk. The disease has a predilection for women (2 : 1). Up to 65% of patients with cutaneous LP will manifest concurrent oral lichen planus (OLP), and oral lesions comprise the sole manifestation of LP in approximately 15–35% of cases. OLP affects an estimated 0.1–4% of the population.
Clinical Features
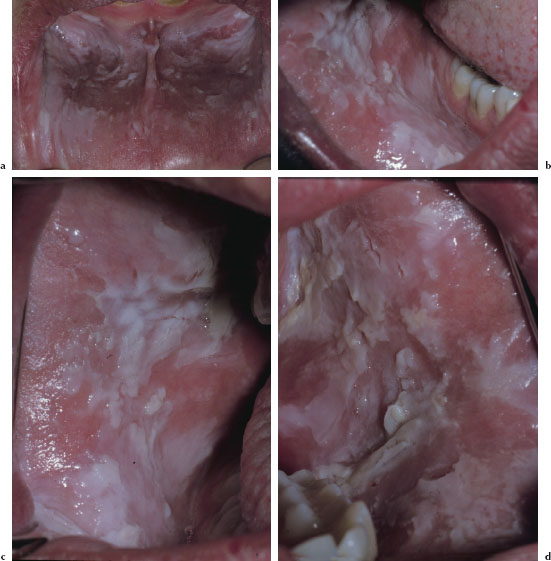
There are at least three recognized forms of OLP: reticular, atrophic, and erosive. The reticular variant is thought to be the most common form and is characterized by mucosal keratotic lines, plaques, or papules arranged in a characteristic lacy pattern of Wickham striae (Figures 5.18a–5.18f). The atrophic form is a combination of the reticular form and erythematous component (Figure 5.19), while the erosive form is a combination of the reticular form and a shallow ulcerative component (Figure 5.20a). All forms of LP may be accompanied by skin lesions (Figure 5.20b). Reticular lesions tend to be asymptomatic, while atrophic and erosive forms are likely to be painful. The most commonly affected sites are the buccal mucosa, tongue, lips, floor of the mouth, palate, and gingivae. OLP typically presents in a bilateral and symmetrical fashion. Patients often present with a variable mix of all three forms, and in some cases the characteristic striae may be absent. OLP may wax and wane, but seldom undergoes total remission.
Figures 5.18a–f. Lichen planus.
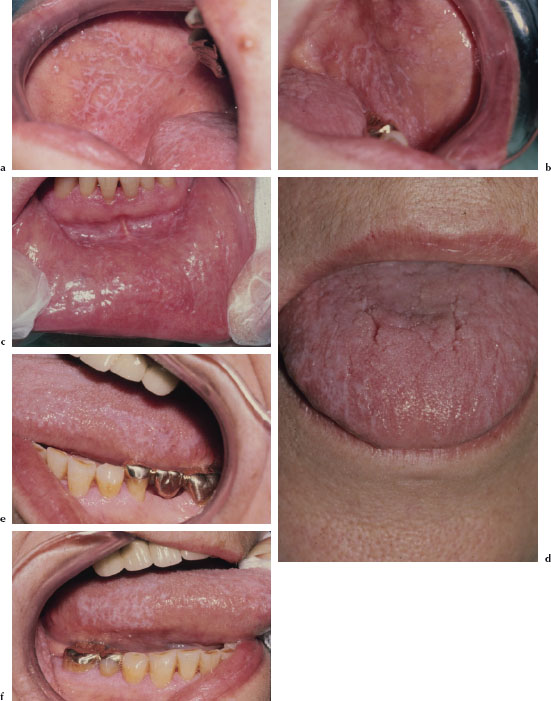
Figure 5.19. Lichen planus.
Figures 5.20a and b. Lichen planus.
Diagnosis
A working diagnosis of LP is usually established when the characteristic striae are present. A confirmatory biopsy should always be obtained and is essential in order to diagnose equivocal cases. The variable appearance of OLP results in an extensive differential diagnosis including oral candidiasis, lichenoid drug reactions, erythema multiforme, epithelial dysplasia, mucous membrane pemphigoid, pemphigus vulgaris, oral cancer, periodontal disease, graft-versus-host disease, lupus erythematosus, gastrointestinal disease, anemia, leukoplakia, erythroplakia, and linear IgA disease.
Treatment
There is no cure for OLP, and treatment is based on symptoms. Asymptomatic reticular OLP requires no treatment. Symptomatic atrophic or erosive forms of OLP often respond well to topical steroids, while more severe disease may require systemic therapy. In all cases, OLP should be routinely monitored, as patients with OLP appear to be at an increased risk of developing oral cancer.
White Sponge Nevus
White sponge nevus is an uncommon benign hereditary disorder that affects the mucous membranes.
Clinical Features
White sponge nevus is characterized by the presence of folded or corrugated white plaques. Symmetrical involvement of the buccal mucosa is invariably observed (Figures 5.21a–5.21d.), but other oral mucosal sites, along with the nasal, vaginal, and rectal mucosa, may be affected.
Figures 5.21a–d. White sponge nevus.
Diagnosis
The clinical appearance, combined with a family history of disease, makes for an easy diagnosis.
Treatment
No treatment is necessary for this benign condition.
Snuff Keratosis
Snuff keratosis represents the reactive mucosal response to the placement of smokeless tobacco products. The smokeless tobacco, along with numerous other ingredients within the smokeless tobacco, acts to irritate the oral mucosal membranes. While there is a tangible risk of dyplasia occurring in snuff keratosis, recent epidemiological data suggests the prevalence of oral epithelial dysplasia in such lesions is generally low. While the rate of malignant transformation is low and the process is slow, the relative risk for carcinoma of the buccal/labial mucosa and gingivae among female chronic users is 50 times greater than among nonusers. It is estimated that 7.7 million people used smokeless tobacco products in 2003.
Clinical Features
Snuff keratosis presents as a fairly characteristic corrugated or folded grayish-white plaque affecting the labial and vestibular mucosa (Figures 5.22, 5.23, 5.24, and 5.25). These lesions develop in those areas where the smokeless product is routinely placed. Most lesions are fairly well localized, but those associated with extensive smokeless tobacco use may be widespread.
Diagnosis
The clear association between smokeless tobacco placement and lesion development makes for a straightforward diagnosis.
Figure 5.22. Snuff keratosis.
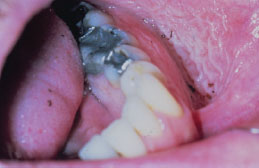
Figure 5.23. Snuff keratosis.
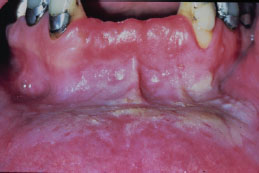
Figure 5.24. Snuff keratosis.
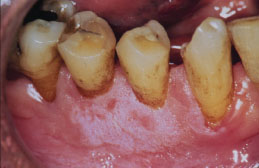
Figure 5.25. Snuff keratosis.
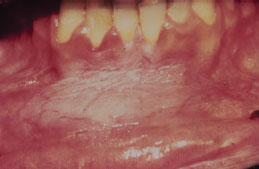
Treatment
Any patient with snuff keratosis should be encouraged to quit using smokeless tobacco and should be advised of available cessation resources. Discontinuation of the smokeless tobacco usually results in lesion resolution. A biopsy is rarely necessary, but is indicated to assess any area of snuff keratosis that is clinically suspicious or unusual looking.
Amalgam Tattoo
An amalgam tattoo is a well-circumscribed, flat pigmented oral lesion. They arise when small particles of dental amalgam, composed primarily of silver (Ag) and mercury (Hg), are inadvertently implanted into the oral soft tissues during dental procedures. It has also been suggested that they may develop in the oral soft tissues as a result of chronic exposure to restorative materials containing Hg, Ag, and sometimes copper, zinc, and tin. Amalgam tattoos are common, affecting about 8% of the population.
Clinical Features
An amalgam tattoo typically presents as an asymptomatic, solitary gray, blue, or black macule on the oral mucosa. They most commonly occur on the gingivae, alveolar ridge (Figure 5.26a), cheek, floor of the mouth, palate, and tongue. Most lesions are less than 1 cm in size. In some cases, evidence of small radiopaque amalgam flecks may be observed on a routine radiograph (Figure 5.26b).
Diagnosis
The diagnosis of an amalgam tattoo is typically based on the characteristic clinical findings combined with the history of prior dental work. A biopsy is recommended for equivocal cases. Other lesions to consider in the differential diagnosis include physiologic pigmentation, an oral melanotic macule, nevi, melanoma, Kaposi’s sarcoma, pigmentation associated with systemic disease (e.g., Addison’s disease, Cushing’s syndrome), and disorders related to blood or blood vessels.
Treatment
An amalgam tattoo does not require treatment. However, all pigmented oral lesions should be viewed with suspicion and if the diagnosis is questionable, a biopsy should be performed. For lesions deemed cosme/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


