Hereditary, Developmental, and Environmental Influences on the Formation of Dentofacial Deformities
• Development of the Head and Neck
• Syndromes and Anomalies that Include Dentofacial Deformities
• Hereditary Tendencies in the Development of Dentofacial Deformities
• Environmental and Neuromotor Effects on Skeletal Growth
• Effects of Trauma on Skeletal Growth
The term dentofacial deformity is generally used to describe a significant disproportion of the jaws in association with malocclusion. It is estimated that, at a minimum, 5% of the population will have a discrepancy that falls into the developmental dentofacial deformity category. Another 45% of the population will fall into a much larger group of those with malocclusion but without what is considered a “notable” jaw discrepancy component.2,3,5,56,65,72 After jaw growth is complete (i.e., 14 to 16 years of age in girls and 16 to 18 years of age in boys), the definitive reconstruction of a dentofacial deformity can go forward.
Development of the Head and Neck
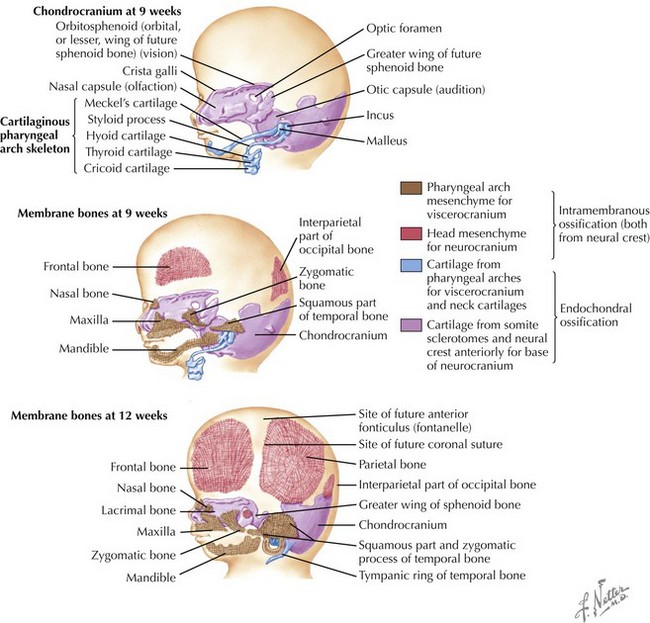
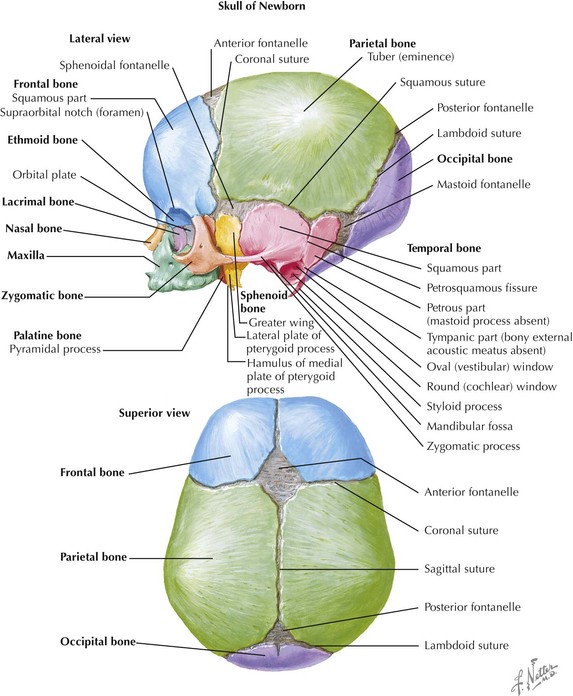
Skull
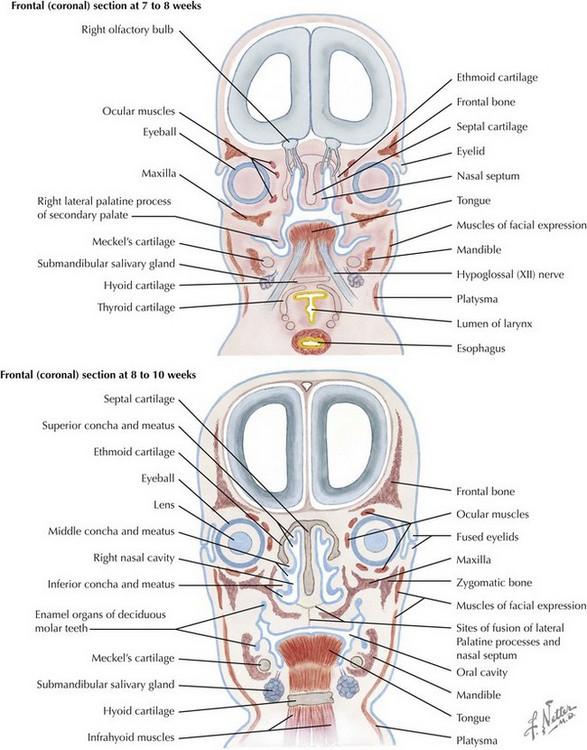
The skull is formed from the lateral plate mesoderm (the neck region), the paraxial mesoderm, and the neural crest (Fig. 4-1). The bony skull is formed by one of two mechanisms: intramembranous ossification or endochondral ossification. Skull development is divided into two parts: the viscerocranium and the neurocranium. The viscerocranium forms the bones of the face, whereas the neurocranium forms the bones of the cranial base and the cranial vault. The neurocranium can be divided into the membranous neurocranium and the cartilaginous neurocranium. The anterior fontanel (bregma) has a wide range of expected closure between 4 and 26 months of age. The posterior fontanel (lambda) generally closes between 1 and 2 months of age.
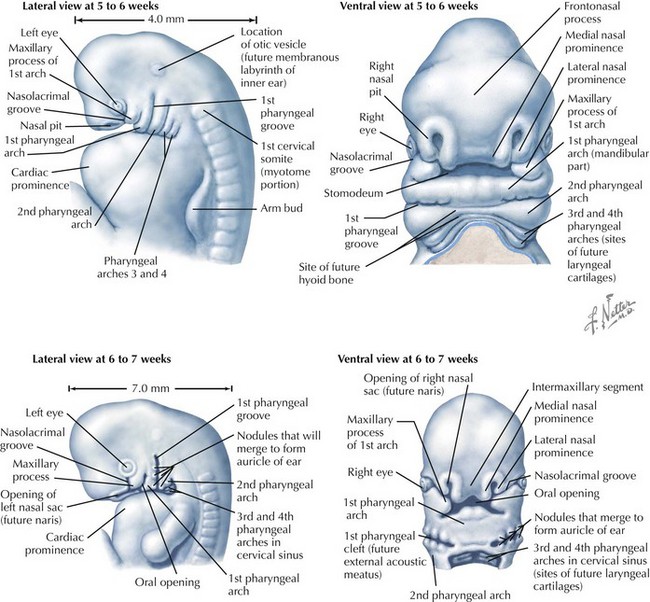
Face
The face is formed mainly from the neural crest, which makes three “swellings” that surround the stomodeum (Fig. 4-2). The three swellings are the frontonasal prominence, the maxillary prominence (from the first pharyngeal arch), and the mandibular prominence (from the first pharyngeal arch).
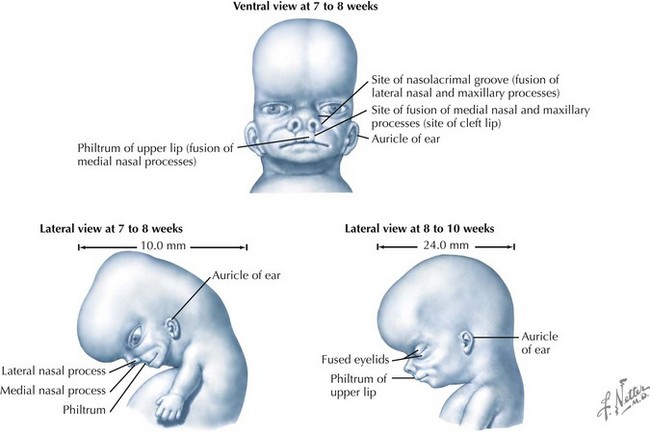
Palate
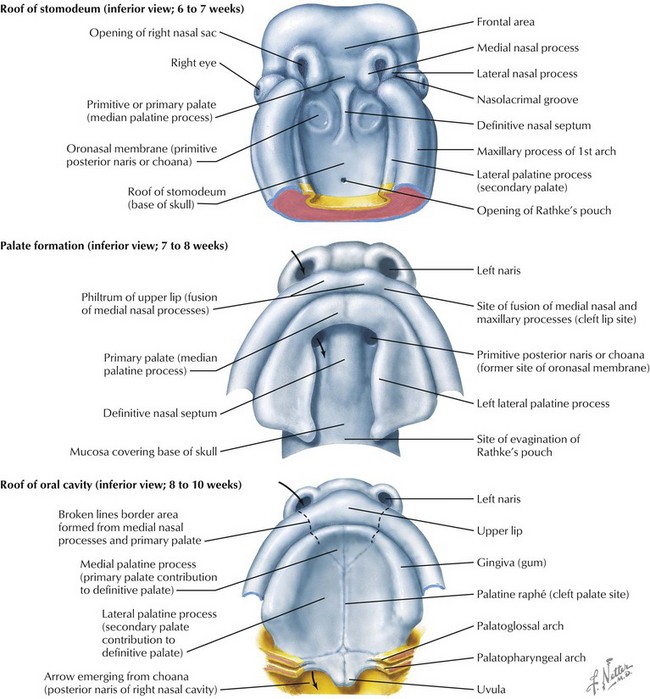
The palate is formed by the primary palate (intermaxillary segment) and the secondary palate (protrusions from the lateral prominences) (Fig. 4-3). The intermaxillary segment (primary palate) is the initial portion of the palate to develop. It contains the central and lateral incisors. Swellings of the maxillary prominence form shelves that project medially but that are separated by the tongue. When the tongue no longer occupies the space between the palatal shelves, these processes fuse together to form the secondary palate. The primary and secondary palatal tissues all meet at the incisal foramen. The primary and secondary palates and the nasal septum fuse to form the definitive palate.
Syndromes and Anomalies that Include Dentofacial Deformities
Many of the syndromes and anomalies that involve the cranio-orbito-zygomatic (upper face) region also affect the maxillomandibular (lower face) location.1,20,124 These conditions may involve any of the tissue components, including the skin, the muscles, the nerves, the fat, the vessels, the bone, the teeth, the cartilage, and the associated viscera (i.e., brain, ears, eyes, salivary glands, sinuses). Known syndromes and anomalies comprise only a small proportion of the dentofacial deformity group (see Chapters 27 through 31).19 These conditions can be further subdivided according to etiology or tissue of origin.
Deficiencies of Midline Tissue Structures
Holoprosencephalies involve variable deficiencies of midline tissues. These structural deficiencies have been documented to occur early during embryologic development.16,18,23,27 They may range from severe brain anomalies to simple absence or hypoplasia of the corpus callosum. Severe facial anomalies such as cyclopia, ethmocephaly, and cebocephaly are always associated with severe holoprosencephaly, and they are incompatible with life. This can also be true of premaxillary agenesis; however, in some instances, patients may survive for several months or even years. There are reported cases of premaxillary agenesis with normal head circumference and no brain abnormalities (i.e., without holoprosencephaly), although these patients sometimes have mild mental deficiencies. Syntelencephaly (the absence or hypoplasia of the corpus callosum or absent or hypoplastic olfactory tracts and bulbs) is compatible with life and should thus be treated. Associated facial anomalies include retinal colobomas, cleft lip, cleft palate, single maxillary central incisor, and midface deficiencies. Holoprosencephaly has multiple causes and includes many chromosomal types, many monogenetic disorders, and at least three teratogens.
Deficiencies and Anomalies of Neural Crest Origin
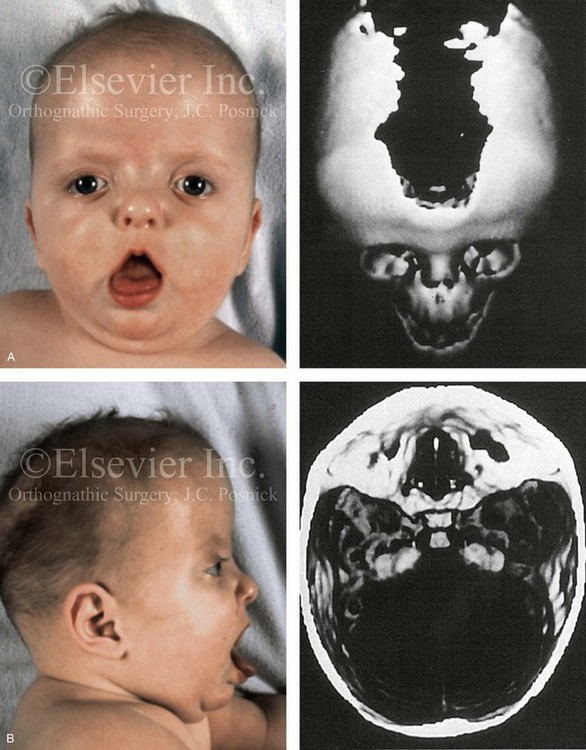
Most of the tissues of the face, including the muscular and skeletal components, are derived from the mesoderm. Interestingly, throughout the rest of the body, these components (i.e., muscle and skeleton) are derived from an ectodermal origin.19 The facial mesodermal elements develop from the neural crest and then migrate downward beside the neural tube and laterally under the surface ectoderm. When the neural crest cells have completed migration to their specific location, then facial development is dominated by specific growth centers, the formation of visceral structures (e.g., brain, eyes, sinuses), and the differentiation of the tissue layers. Facial anomalies of neural crest origin are thought to arise when the neuroepithelium has apoptosis (i.e., the cells die).25 This results in less neuroepithelium to migrate through into the facial locations and to ultimately differentiate. Current knowledge of Treacher Collins syndrome indicates that this is not a migration problem but simply a lack of cells to migrate (Figs. 4-4 and 4-5).
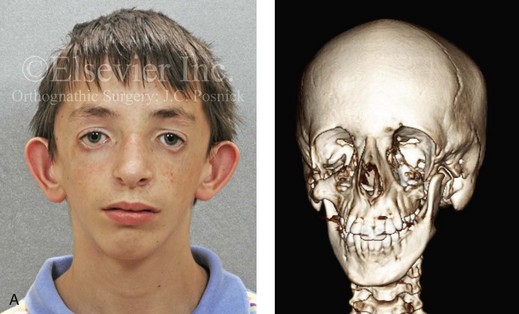
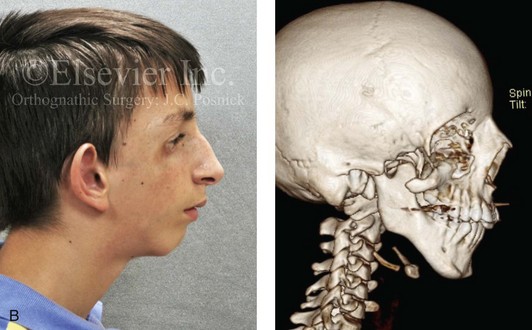
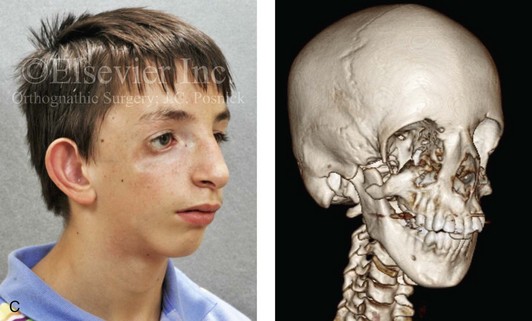
Figure 4-4 An 11-year-old boy who was born with Treacher Collins syndrome has typical malformations of the soft tissues and the skeletal structures within the first and second branchial arches on both sides. There is significant hypoplasia of each zygomatic complex, the orbits, and the maxillomandibular structures (Kaban type II-A mandible). The soft tissues of the adnexal region are deficient. The external ears are malformed, but all parts are present. The central part of the face is fully formed. A, Frontal facial and computed tomography scan views. B, Profile facial and computed tomography scan views. C, Oblique facial and computed tomography scan views.
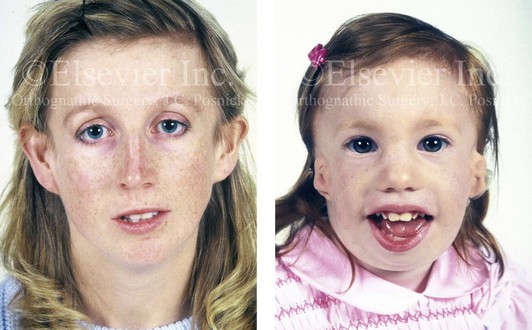
Figure 4-5 This mother and daughter demonstrate the extent of variation of expression of Treacher Collins syndrome within a family. The mother was not aware that she carried the Treacher Collins gene until after the birth of her daughter. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment. J Oral Maxillofac Surg 55:1120–1133, 1997.
Teratogens that are known to result in head and neck (neural crest) congenital anomalies include cytomegalovirus (microcephaly, hydrocephaly, microophthalmia); Dilantin (cleft lip and palate); vitamin D excess (premature suture closure); Valium (cleft lip and palate); Rubella virus (microophthalmia, cataracts, deafness); and thalidomide (variations of hemifacial microsomia and Treacher Collins syndrome). Despite these known relationships, teratogenic agents are not the most frequent causes of these syndromes.1
The branchial arch syndromes are neural crest anomalies that comprise an etiologically heterogeneous group of disorders.14 They account for only a small percentage of the patients who present to the surgeon or orthodontist in need of jaw reconstruction. The best known of these are hemifacial microsomia, its variant Goldenhar syndrome, and Treacher Collins syndrome.14 Hemifacial microsomia affects aural, zygomatic, and mandibular growth at a minimum (Fig. 4-6). The disorder may be mild or severe, and it is generally limited to one side of the face. However, bilateral involvement, with more severe expression on one side, is also known to occur. Goldenhar syndrome, which is a variant of hemifacial microsomia, also includes epibulbar dermoids and vertebral anomalies. Involvement is often but not always limited to the face. There may be cardiac, renal, skeletal, and central nervous system anomalies (see Chapter 28). At a minimum, patients with Treacher Collins syndrome demonstrate the physical findings of bilateral zygomatic hypoplasia, down-slanting palpebral fissures, malformed ears, and micrognathia (see Chapter 27). Inheritance occurs in an autosomal-dominant fashion, and expressivity varies (see Figs. 4-4 and 4-5). The syndrome maps to 5q32-q33.1, and mutations in the TCOF1 gene are of the nonsense, insertion, deletion, or splice-site types.14,30,139
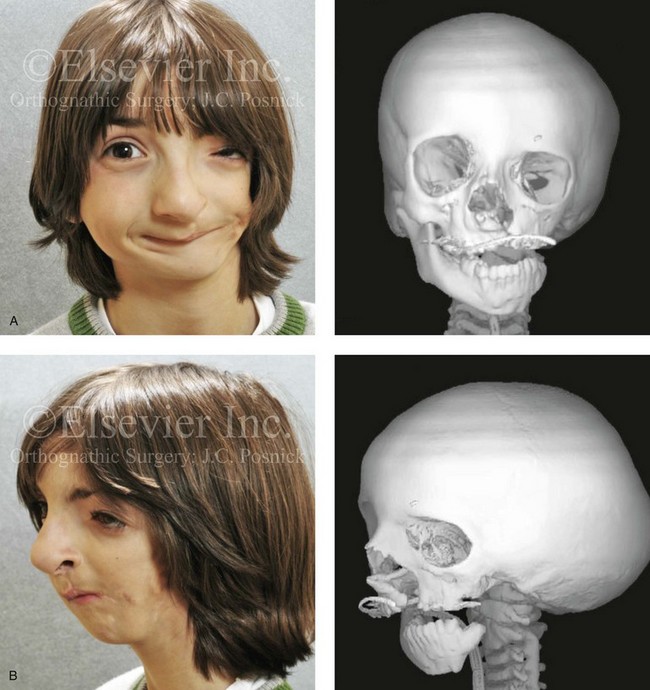
Figure 4-6 A 7-year-old boy born with hemifacial microsomia and unilateral cleft lip and palate (left side) has typical malformations of the soft tissues and skeletal structures within the first and second branchial arches on the left side. There is an absence of the left zygomatic complex and hypoplasia of the orbit, the anterior cranial vault, and the maxilla. The mandibular malformation is a Kaban type III. The soft tissues of the left external ear, the ear canal adnexal structures, and the cheek region are markedly deficient. A, Frontal facial and computed tomography scan views. B, Oblique facial and computed tomography scan views.
Clefting of the Lip and Palate
The most prevalent congenital defect of dentofacial development is clefting of the lip, the palate, or both (Figures 4-7 through 4-19). This condition occurs in approximately 1 in 1000 whites, 1 in 500 Asians, and 1 in 2000 blacks.27 The etiology of clefts is often complex and multifactorial. Evidence supports the view that genetic factors are associated with orofacial clefting.37 In twins with cleft lips and palates, concordance is far greater among monozygotic twins (40%) as compared with dizygotic twins (4.2%). In twins with isolated cleft palate, concordance is also higher among monozygotic twins (35%) as compared with dizygotic twins (7.8%).138 Nevertheless, orofacial clefting is heterogeneous and variable, and it is likely determined by a number of major genes, minor genes, environmental factors, and a developmental threshold.37 Some syndromes associated with clefting in which specific gene mutations have been identified include van der Woude syndrome (IRF6), popliteal pterygium syndrome (IRF6), autosomal-recessive cleft palate/ectodermal dysplasia (PVRL1), hypodontia/clefting (MSX1), Hay-Wells syndrome (TP63), and cleft palate/ankyloglossia (TBX22). These mutations explain only about 5% of clefting cases, with an additional 10% to 15% of cases explained by variations involving the IRF6 gene.110 Although orofacial clefting is caused by a malformation, the development of secondary deformities of the jaws after birth is well known (see Chapters 32, 33, and 34). For the most part, maxillary hypoplasia in patients with orofacial clefts is felt to be the result of palate surgical interventions carried out during infancy and early childhood and not from the primary congenital anomaly. Although van der Woude and Stickler are just 2 of more than 100 clefting syndromes, they demonstrate the importance of having an awareness of associated anomalies to assist with family counseling and to achieve effective reconstruction.
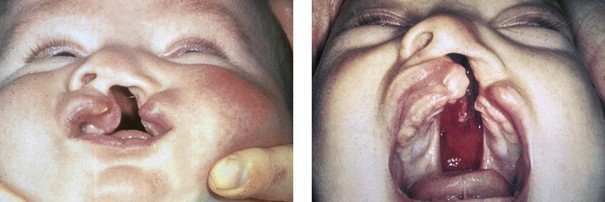
Figure 4-7 Cleft lip and palate represents a spectrum of morphologic findings that are initially dependent on the individual’s specific anomalies but that are then affected by surgical intervention during growth. A newborn with a complete (left side) unilateral cleft lip and palate is shown.
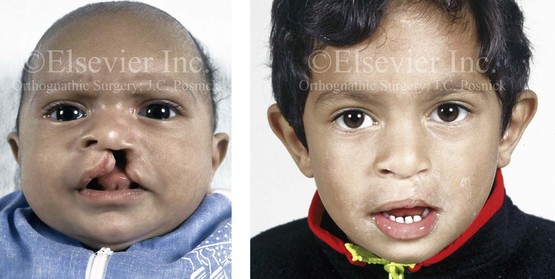
Figure 4-8 A Hispanic child was born with complete clefting of the left primary lip and palate. He was adopted when he was approximately 1 year old, before repair. He then underwent single-stage primary lip, nasal, and palate repair. He is shown before repair and then at the age of 2 years after single-stage repair.
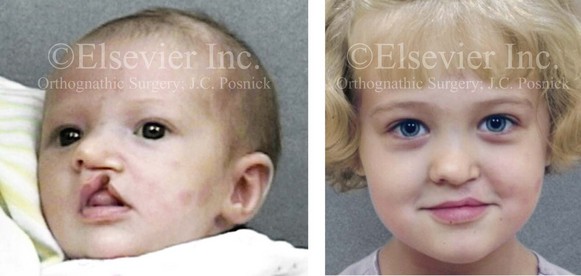
Figure 4-9 A child was born with an incomplete cleft of the left lip that also involved the alveolar ridge back to the incisal foramen. She is shown before primary lip and nasal reconstruction and then at the age of 5 years before alveolar cleft grafting.
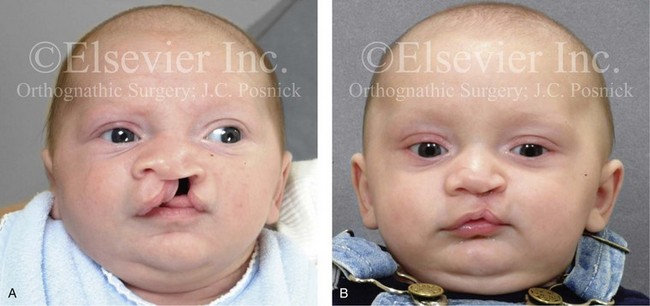
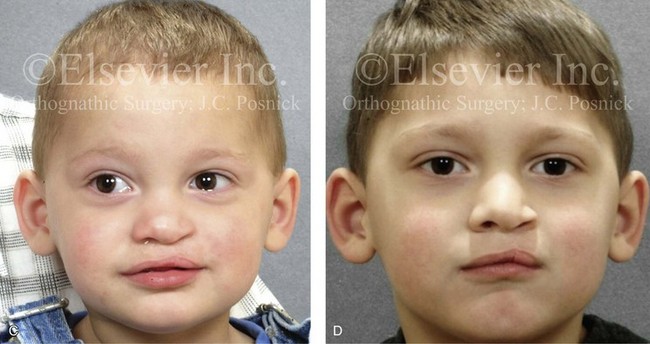
Figure 4-10 A child was born with a complete (left side) unilateral cleft lip and palate. He is shown before and at intervals after primary lip and palate repair. A, He is shown before primary lip and nasal reconstruction and B, at the age of 10 months, just before cleft palate repair. C, The same child at the age of 3 years and D, at the age of 8 years just before mixed dentition bone grafting.
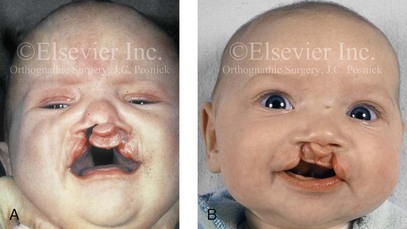
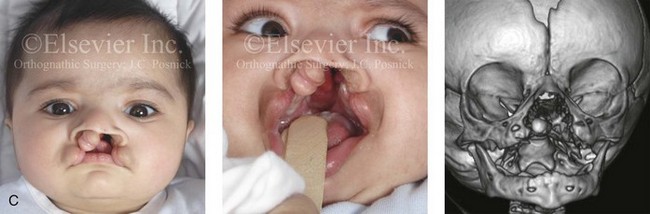
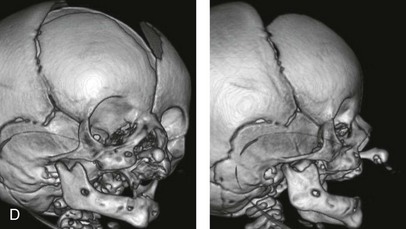
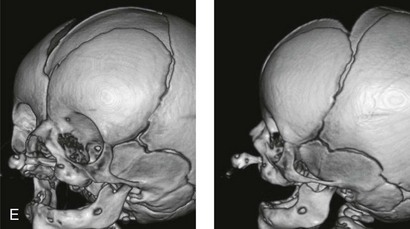
Figure 4-11 Three newborns with bilateral cleft lip and palate (BCLP) are shown. A, A newborn with complete BCLP. B, A newborn with BCLP but with partial attachment of the lip and the nasal floor on the left side. C, A 3-month-old infant with complete BCLP is shown. The premaxilla is attached to the septum of the nose; it is forwardly projecting without attachment to the lateral lip segments. The wide separation of the palatal shelves is also demonstrated by the intraoral view. D and E, A series of computed tomography scan views of the maxillofacial complex of the infant shown in C demonstrate the skeletal anatomy associated with BCLP.
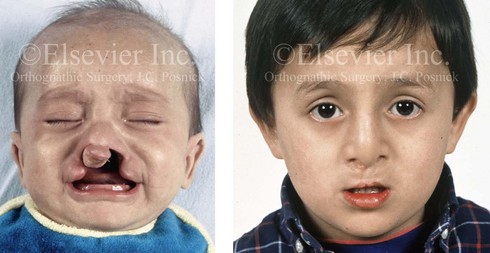
Figure 4-12 A child born with complete bilateral cleft lip and palate. He is shown before and then 7 years after primary lip and palate repair, just before mixed dentition bone grafting.
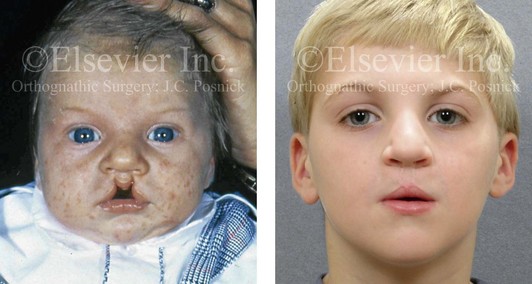
Figure 4-13 A child born with incomplete bilateral cleft lip and palate. He is shown before and then 8 years after primary lip and palate repair, just before mixed dentition bone grafting.
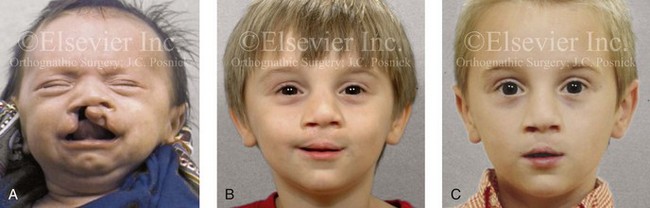
Figure 4-14 A child born with a complete cleft of the right lip and palate and incomplete clefting of the left lip. He is shown A, before and then B, at 2 and C, 8 years after primary lip and palate repair. The last photo was taken just before mixed dentition bone grafting.
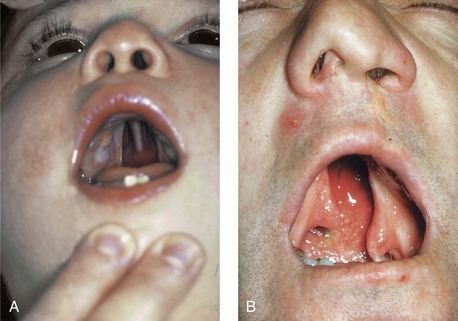
Figure 4-15 A, A child born with complete clefting of the secondary palate (i.e., incisal foramen through the uvula) is shown. B, An adult born with a complete unilateral cleft lip and palate is also shown. He underwent lip repair during childhood, but the cleft palate was neglected. Note the severe septal deviation that obstructs the right nasal valve.
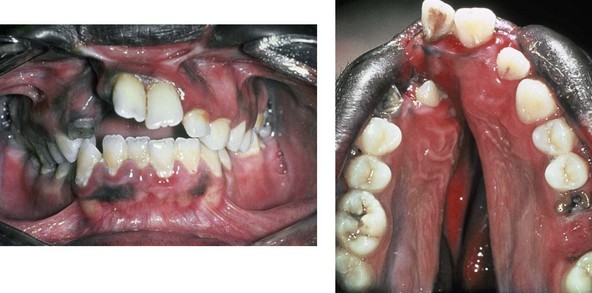
Figure 4-16 A teenage boy from Jamaica who was born with a bilateral cleft lip and a unilateral cleft of the alveolar ridge and palate. He underwent rudimentary lip repair as a child. The unrepaired clefted alveolus and palatal anatomy are shown. Note the normal growth parameters of the upper jaw when the palate is unrepaired. With a lack of continuity of the alveolar ridge, there has been typical distorted growth of the maxilla.
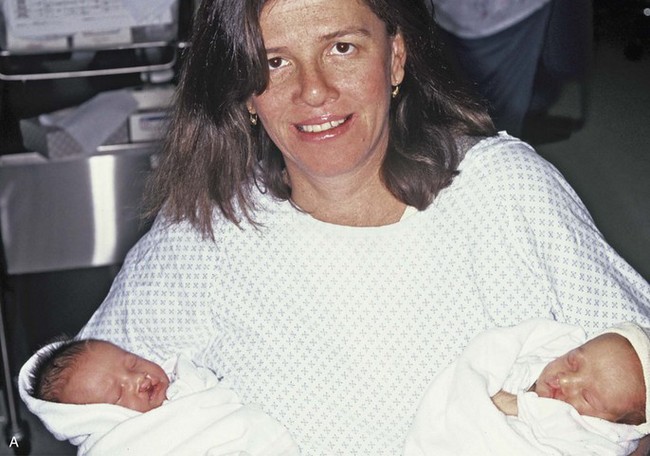
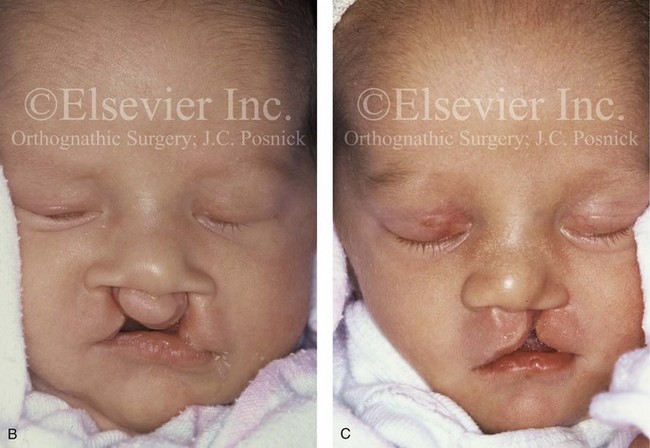
Figure 4-17 A woman who was born with bilateral cleft lip and palate and pitting of the lower (central) lip. At the time of her pregnancy, an ultrasound confirmed twins, one of which was suspected of having bilateral cleft lip and palate and the other of having unilateral cleft lip and palate. This was documented at the time of delivery. A, A family with van der Woude syndrome, including a mother with a repaired bilateral cleft lip and palate and newborn twins. B, Twin A with bilateral cleft lip and palate. C, Twin B with unilateral cleft lip and palate.

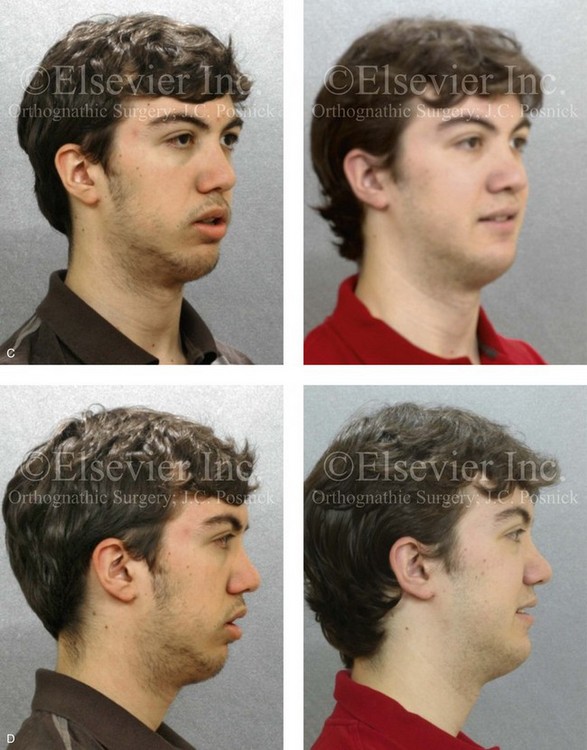
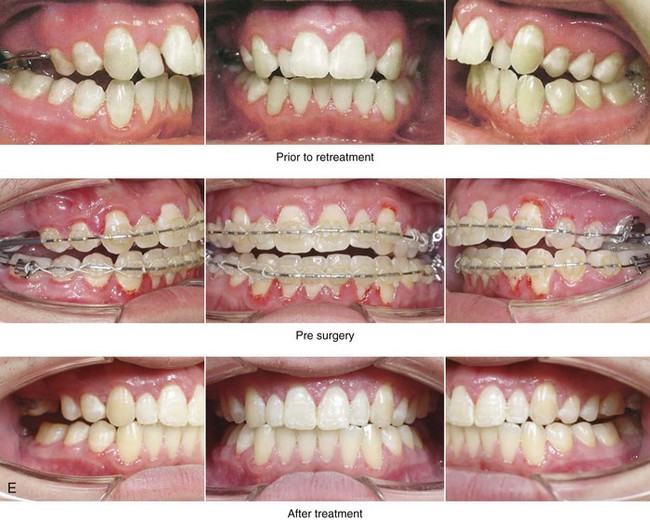
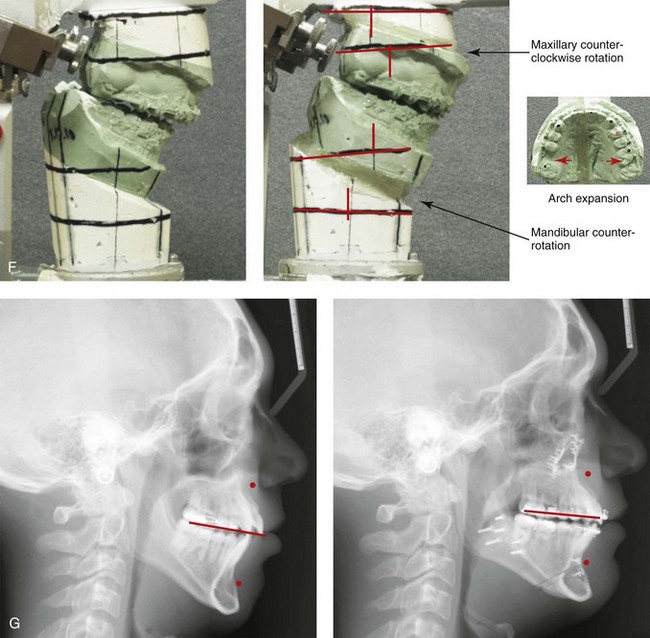
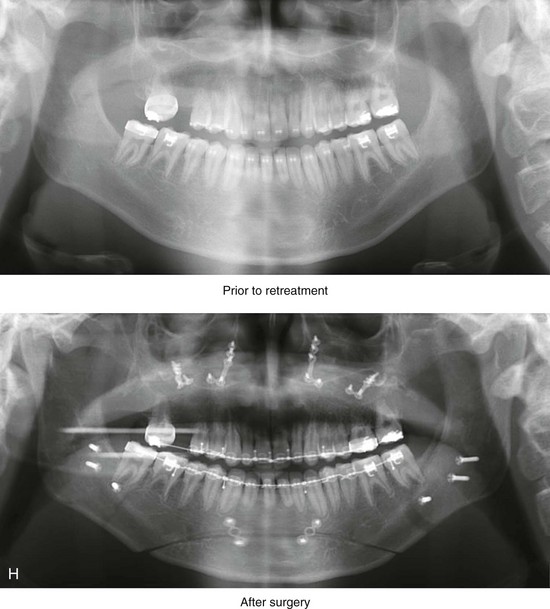
Figure 4-18 A 16-year-old boy who was born with Stickler syndrome (type II collagen mutation). At the time of birth, Pierre Robin sequence was appreciated. The patient underwent repair of the cleft palate before he was 1 year old. He had positive eye findings, and he required the treatment of a retinal detachment during his teenage years. A small cataract is being followed. He arrived for the evaluation of a jaw deformity and malocclusion characterized by maxillomandibular deficiency with anterior open-bite malocclusion. This was combined with chronic obstructive nasal breathing and a long face growth pattern. Attempted growth modification and camouflage orthodontics earlier during life were ineffective. There was generalized root deficiency throughout the maxillary and mandibular dentition, likely as a result of the collagenopathy. A, Frontal views in repose before and after treatment. B, Frontal views with smile before and after treatment. The patient agreed to an orthodontic and surgical approach. Further orthodontic (dental) decompensation was cautiously carried out as a result of the compromised periodontal apparatus. The procedures included maxillary Le Fort I osteotomy (vertical intrusion, horizontal advancement, counterclockwise rotation, and arch expansion); bilateral sagittal split ramus osteotomies (horizontal advancement and counterclockwise rotation); osseous genioplasty (vertical shortening and horizontal advancement); and septoplasty, inferior turbinate reduction, and nasal floor recontouring. C, Oblique facial views before and after treatment. D, Profile views before and after treatment. E, Occlusal views before retreatment, with orthodontics in progress, and after treatment. F, Articulated dental casts that indicate analytic model planning. G, Lateral cephalometric radiographs before and after treatment. H, Panorex radiographs before and after treatment that demonstrate generalized limited root formation. The right maxillary first molar was lost 5 years earlier as a result of limited roots. The right maxillary first molar was lost 1½ years after surgery as a result of limited root support; the right mandibular first molar will likely be lost to similar pathology. Periodontal evaluation and treatment are ongoing. Future plans are for the placement of dental implants in the right posterior maxilla and mandible.
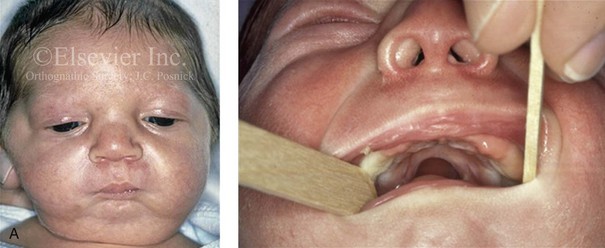
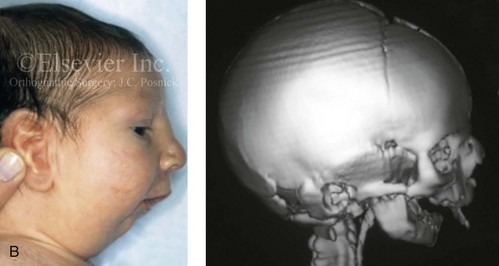
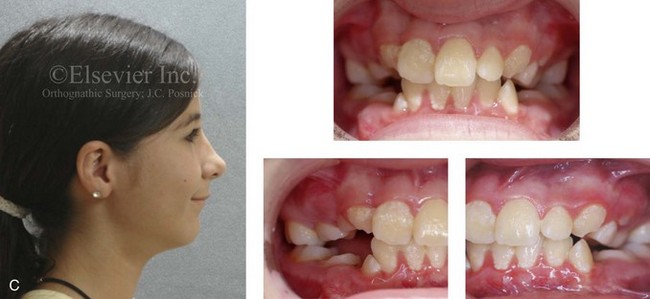
Figure 4-19 A child born with Robin sequence, which consists of retrognathia with glossoptosis, clefting of the secondary palate, and a degree of respiratory distress. A, Frontal facial and intraoral views demonstrating clefting of the secondary palate. B, Profile facial view indicating retrognathia. Computed tomography scan demonstrating retrognathia but with all of the components of the jaw present. For this child, the small mandible is the result of deforming forces during fetal development rather than the result of a malformation. Catch-up growth of the mandible is anticipated during childhood. C, Profile and occlusal views when the patient was 9 years old, with expected catch-up growth.
Van der Woude Syndrome
In 1845, Demarquay was the first to report the occurrence of congenital sinuses of the lower lip in combination with cleft lip and palate; this condition became known as van der Woude syndrome. This syndrome occurs in roughly 1% to 2% of patients with lip and palatal clefts. Van der Woude syndrome has an autosomal-dominant inheritance pattern; it has approximately 90% penetrance, and it is variably expressed (see Fig. 4-17). Manifestations of the syndrome outside of the oral and facial regions are unusual. Generally, the pits are characterized as depressions that are observed on the vermilion border of the lower lip; they are usually bilateral and symmetrically placed, although an asymmetrical pit or single central pit may occur. Roughly 33% of patients with van der Woude syndrome have pits without clefting, 33% have pits with cleft lip and palate, and 33% have lip pits with isolated cleft palate or submucous cleft palate. Approximately 10% of those individuals with van der Woude syndrome do not exhibit lip pits.
Stickler Syndrome
In 1967, Stickler and Pugh were the first to describe a series of patients who had a combination of eye findings, hearing loss, isolated cleft palate, a Marfanoid habitus, and long-bone changes.1 It is now known that Stickler syndrome is associated with basic defects in collagen. These collagenopathies include mutations on type II collagen and mutations in type XI collagen.25 The inheritance pattern with this syndrome is autosomal dominant with variable expressivity. Cohen and others have reviewed the relationship between high myopia and clefting of the secondary palate and the other features of Stickler syndrome.19 High myopia (i.e., 8 to 18 diopters) is found in 75% of patients by the time they are 5 years old. The eye findings are progressive, with vitreous and chorioretinal degeneration and retinal detachment observed in 70% of patients by the time they are 20 years old. Additional eye findings include astigmatism, cataracts, strabismus, and glaucoma. Individuals with Stickler syndrome may manifest any or all of a range of craniofacial findings, including mild to moderate midface hypoplasia, shallow orbits with eye proptosis, epicanthal folds, a flat nasal bridge, and submucous or full clefting of the secondary palate. Some affected individuals will have progressive degeneration of the dental roots and the associated periodontium. This is primarily due to the inborn error in collagen production, but it may be exacerbated by malocclusion (secondary trauma) and orthodontic manipulations (see Fig. 4-18). Progressive neurosensory high-tone hearing loss has been reported in 80% of patients. Some patients have a Marfanoid body habitus, whereas others have short stature. There may be progressive early joint degeneration by the mid-adult years. Any newborn who is found to have Robin sequence should undergo an initial genetic and ophthalmologic assessment to rule out Stickler syndrome.
Robin Sequence
Robin sequence is commonly defined as cleft palate, micrognathia, and glossoptosis (see Fig. 4-19). This sequence of events can occur as a result of a variety of etiologic and pathogenetic conditions, and it involves a wide spectrum of phenotypes. The distinction must be made between a malformed mandible (micrognathia) and a deformed mandible that is simply retrognathic. The malformed mandible (i.e., Treacher Collins syndrome) may also be missing parts or sections of the jaw (i.e., Kaban types IIB and III) and cannot be expected to “grow out” and self-correct. This is different from a deformed mandible (i.e., retrognathic at birth) caused by fetal malpositioning, which can be expected to “grow out” and become relatively normal. The newborn’s respiratory compromise that occurs with Robin sequence will vary according to the child’s physical findings, and treatment should proceed accordingly. For example, with spondyloepiphyseal dysplasia congenita, the causes of respiratory compromise include a small and mechanically abnormal chest, tracheobronchial malacia, and central apnea as a result of cervical or medullary compression caused by cervical instability.47 This will require a different level of airway management as compared with an isolated retrognathic mandible, which is simply deformed as a result of intrauterine compression. If the deformed (retrognathic) mandible is present at birth, it is expected to “catch up” during growth and basically to self-correct. For these infants, mandibular osteotomies with advancement carried out during childhood should be avoided.
Achondroplasia
Achondroplasia is the most common condition associated with severe disproportionate short stature.22 The cause of this condition is the failure during development of the primary growth cartilages of the limbs and the cranial base. This results in short arms and legs as well as a characteristic midface deficiency or deformity that is most visually notable at the nasofrontal process.
Most affected individuals do well but attain a final height of only approximately 4 feet. The condition is known to be caused by mutations on the FGFR3 (fibroblast growth factor receptor 3) gene.22 Eighty percent of reported cases are sporadic, whereas 20% are familial. Although craniosynostosis is not a typical feature, three cases with craniosynostosis have been reported.
Premature Suture Closure of the Cranial Vault and Skull Base
The flat bones of the cranial vault develop from mesenchymal condensations over the developing brain (see Fig. 4-1). These bones grow primarily via the apposition of bone at their edges. The regions in which the cranial bony edges collide are called sutures. In addition to appositional growth at the edges (i.e., suture growth), cranial vault development also occurs through the process of remodeling (i.e., resorption and deposition).17 Continued separation of the sutures during the process of the apposition of bone at the edges is also important to the normal growth of the bones of the cranial base and the midface.
Synostosis is a condition of premature fusion (i.e., the arrest of appositional growth at the bone edges) that occurs before the associated visceral structures (e.g., brain, eyes) complete growth.17 When this happens, there is distortion or deformity of the shape of the affected bones and possible compression of the underlying visceral structures (i.e., the brain). Premature closure of the cranial vault sutures (craniosynostosis) that do not extend into the cranial base will have minimal effects on the maxillofacial form (e.g., metopic, unilateral coronal, and sagittal suture synostosis; Figures 4-20, 4-21, and 4-22). However, prematurely fused cranial vault sutures that extend into the cranial base will also affect midfacial growth and development (e.g., Crouzon, Apert, and Pfeiffer syndrome; Figures 4-23 through 4-26; see Chapter 30).
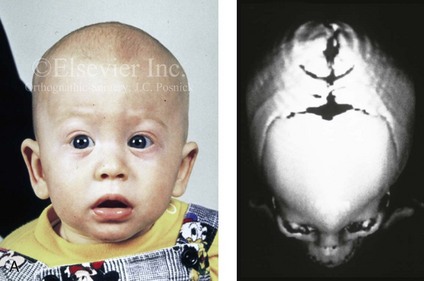
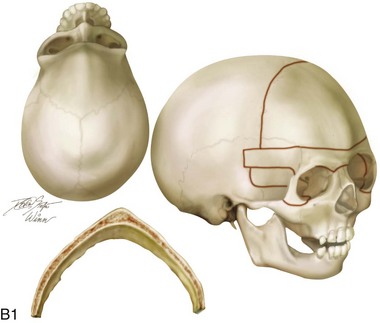
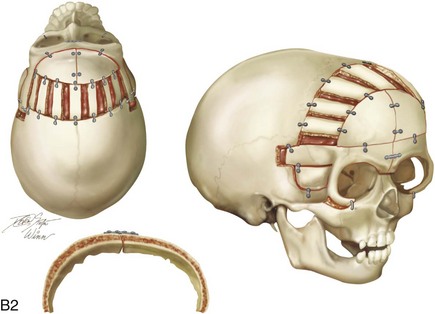
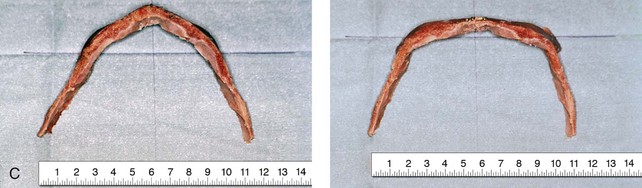
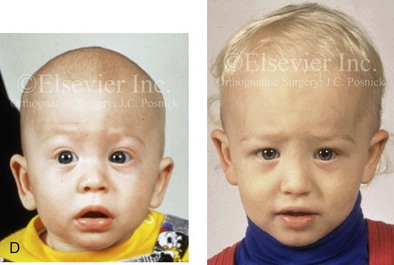

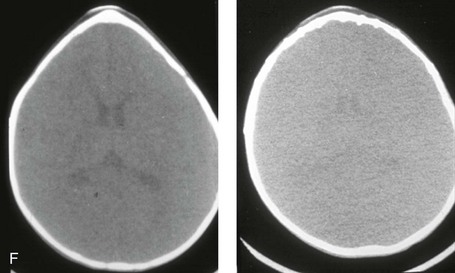
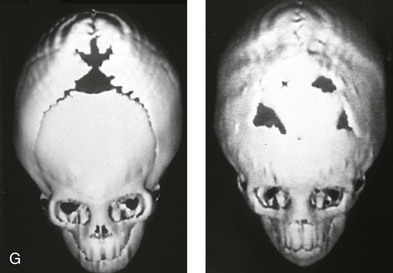
Figure 4-20 A child born with metopic synostosis that resulted in trigonocephaly. He underwent anterior cranial vault and three-quarter orbital osteotomies with reshaping when he was 10 months old. A, Facial and computed tomography scan views before surgery. B, Illustrations of the craniofacial skeleton in a child with metopic synostosis that resulted in trigonocephaly before and after anterior cranial vault and three-quarter orbital osteotomies. C, Bird’s-eye view of removed orbital osteotomy unit before and after reshaping. Part B modified from an original illustration by Bill Winn. D, Frontal views before and 1 year after reconstruction. E, Facial views of patient at 5 years old. F and G, Computed tomography scan views before and after reconstruction.
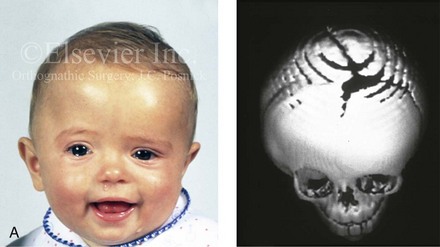
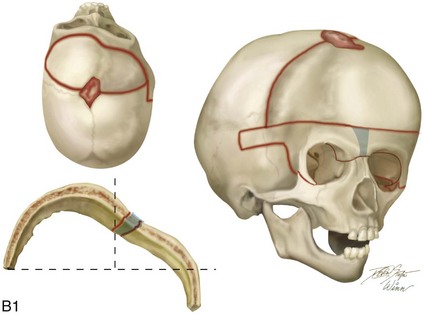
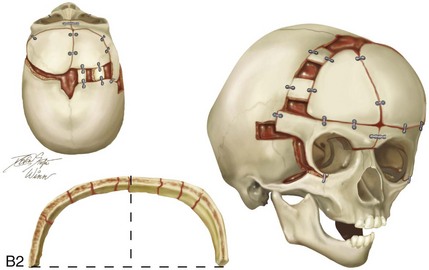
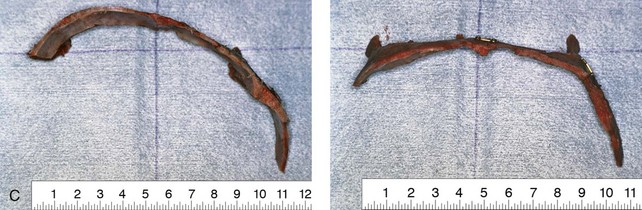
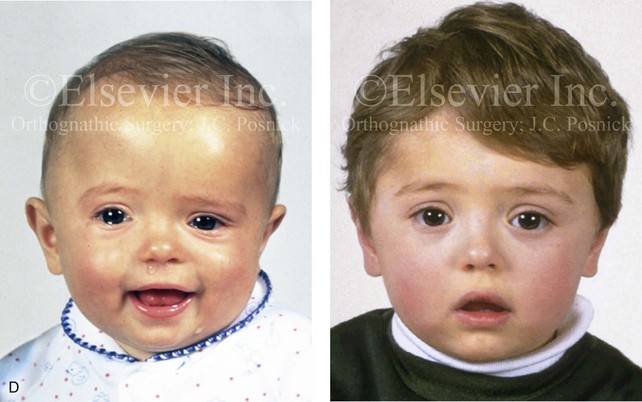
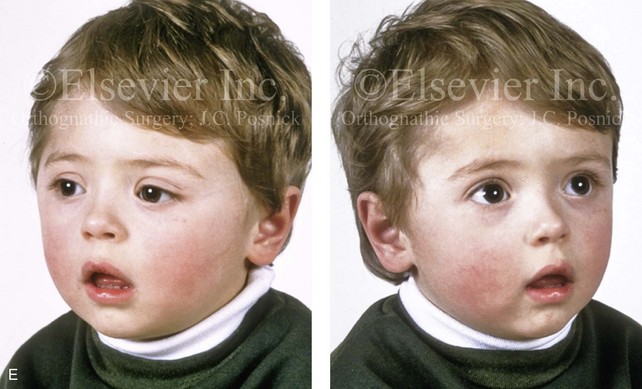
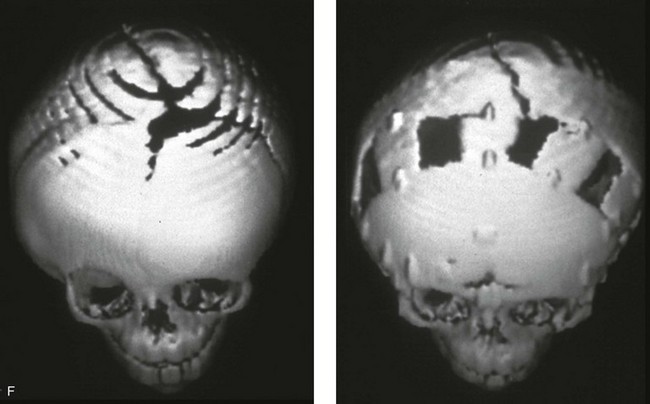
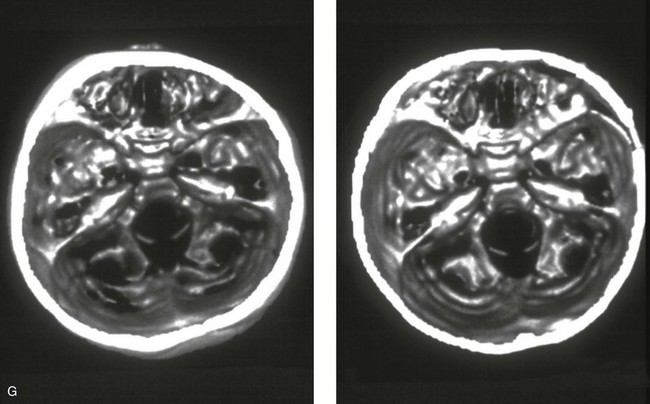
Figure 4-21 A child born with right unilateral coronal synostosis that resulted in anterior plagiocephaly. He underwent anterior cranial vault and three-quarter orbital osteotomies with reshaping when he was 9 months old. A, Facial and computed tomography scan views before reconstruction. B, Illustrations of the craniofacial skeleton in a child with unilateral coronal synostosis that resulted in anterior plagiocephaly before and after anterior cranial vault and three-quarter orbital osteotomies. Part B modified from an original illustration by Bill Winn.C, Bird’s-eye view of removed orbital osteotomy units before and after reshaping. D, Frontal views before and 5 years after reconstruction. E, Oblique views 5 years after reconstruction. F and G, Computed tomography scan views of the cranial vault before and just after reconstruction.
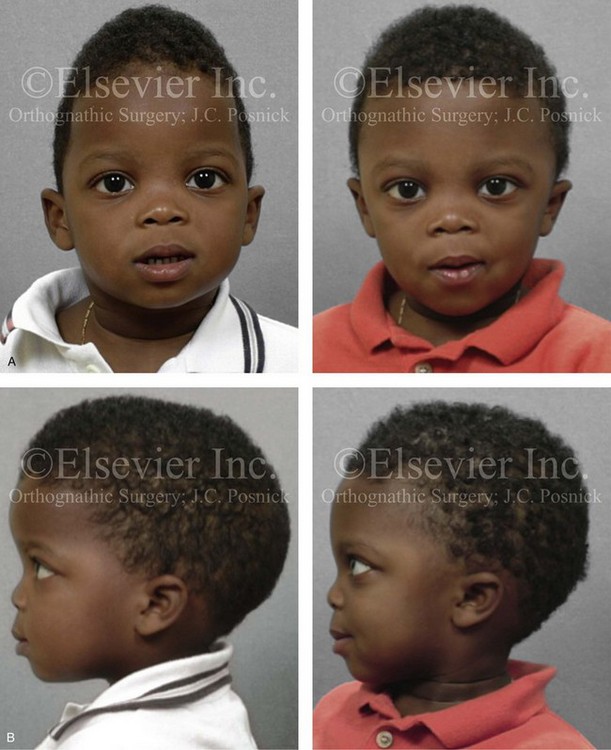
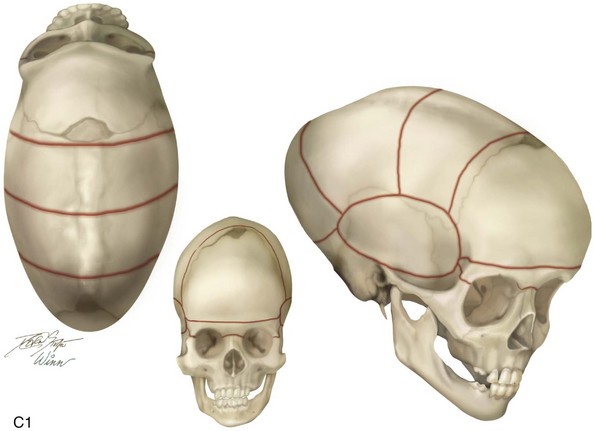
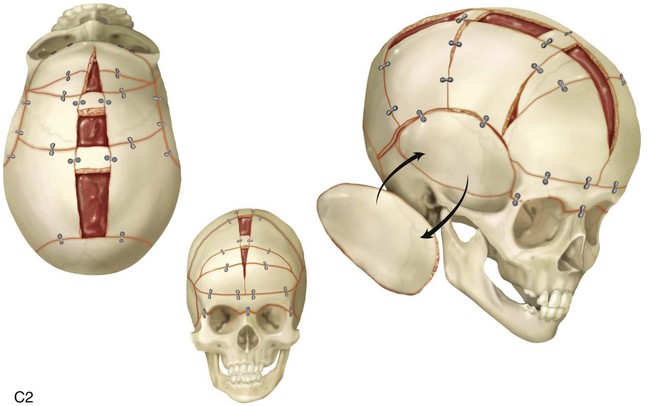
Figure 4-22 A child born with sagittal synostosis that resulted in a scaphocephalic shape of the cranial vault that was characterized by increased anteroposterior length and decreased bitemporal and biparietal width. It was not until the child was 2 years old that he was diagnosed with sagittal synostosis. After a comprehensive evaluation, he underwent total cranial vault reshaping through a coronal (scalp) incision. He is shown before and after a single-stage reconstruction. A, Frontal facial views before and after reconstruction. B, Profile facial views before and after reconstruction. C, Illustrations of the craniofacial skeleton in a child with sagittal synostosis that resulted in scaphocephaly before and after total cranial vault and upper orbital osteotomies with reconstruction. Part C modified from an original illustration by Bill Winn.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


