Selected Complex Malformations that Frequently Require Maxillofacial Reconstruction
Evaluation & Treatment
Klippel–Feil Anomaly
Background
The Klippel–Feil anomaly (KFA) is characterized by the congenital fusion of two or more cervical vertebrae. In its most severe form, there is massive cervical vertebral fusion with a short neck, limited head movement, and a low posterior hairline.2,12,15,21,23,29,37,39,55 Gorlin and Cohen have pointed out that the term vertebral fusion is not accurate because this condition results from the failure of the normal segmentation process.24 Vertebral fusion can be traced to the third embryonic week when the segmentation of the mesodermal somites normally takes place.
KFA was described as early as the 16th century, and similar anatomic findings in the second and third cervical vertebrae have been found in an Egyptian mummy that has been dated to be from about 500 B.C. In 1912, Maurice Klippel and Andre Feil described the postmortem findings of a 46-year-old French tailor who had a short, immobile neck with massive fusion of cervical and upper thoracic vertebrae.34 In 1919, Feil added reports of 13 additional patients, 12 of whose cases had been reported previously.
In 1967, Gunderson and colleagues categorized KFA in accordance with three morphologic types of cervical vertebral fusion.27 Type I consists of the massive fusion of many cervical and upper thoracic vertebrae into bony locks. Type II involves fusion at only one or two interspaces. Type III comprises both cervical fusion and lower thoracic or lumbar fusion.
It is important to remember that KFA is morphologically and etiologically heterogeneous. This was further documented by Gunderson, who used Feil’s classification system to review inheritance patterns.28 Type I cases have almost all been sporadic, with a female predilection. Type II is transmitted as an autosomal dominant disorder. For Type III, an autosomal recessive mode of inheritance has been suggested.
Various authors differ with regard to their interpretation of what constitutes KFA.9,61–63 A malformation with the triad of a short neck, a low posterior hairline, and a painless restriction of cervical motion characterizes just over 50% of patients. The functional limitation of the neck range of motion is the most consistent finding. Despite frequent sparing of the atlanto–axial joint, rotation is typically impaired more than flexion or extension is, with the latter exhibiting a range of more than 90 degrees, even with only one open disc space.
Diverse ocular anomalies are frequently found, with the impairment of extraocular movement being the most frequent.57Convergent strabismus and, less commonly, horizontal nystagmuses are seen. From 25% to as many as 50% of affected children exhibit hearing loss that may be sensorineural, mixed, or conductive.11,16,39,51,60,64 Clefting of the secondary palate is present in 15% to 20% of patients.10,58,59 The combination of hearing loss and cleft palate explains the hypernasality that has been documented in approximately 15% to 20% of patients with KFA. Congenital heart defects occur in 4.2% of patients, which is in stark contrast with the prevalence of 0.6% among all live births in the general population.50 Ventricular septal defect is the most common heart anomaly documented,43 and vascular anomalies may also occur.5,6 Congenital urinary anomalies are a frequent finding, with unilateral renal agenesis occurring in 28% of patients.14 Genital anomalies (e.g., vaginal agenesis) also occur at a higher frequency in these patients than they do in the general population.40,42 Neurologic disturbances that consist of involuntary dyskinesis, spasticity or hyperreflexia, syringomyelia, syringobulbia, disc protrusion, osteophytes, and narrowing at the level of the craniovertebral junction have been reported..3,20,22,36,44,48 Nagib and colleagues found patients with KFA with two unsegmented blocks of bone, cervical stenosis, or cranial involvement to be at highest risk for neurologic disturbances.45–47 Those with only one unsegmented bone were found to be at low risk for cervical injuries. The flared trapezius muscles associated with KFA give the individual an appearance that may be similar to that seen in patients with Turner or Noonan syndrome.8,13,18,38 The cervical vertebral anomalies and other defects of the spine that occur within the oculo–auriculo–vertebral spectrum may also be confused with those of KFA.4,7,8,25,32,33,35,49,52,54,56
Author’s Approach to Facial Reconstruction in Patients with Klippel–Feil Anomaly
The surgical management of KFA requires the accurate diagnosis of each specific problem (see Fig. 1-2).17,26,31 Middle-ear infections should be treated promptly, and any hearing deficit should be aided whenever possible. Clefting of the secondary palate should be repaired in the usual way. Hypernasality as a result of inadequate soft palate motion after initial palate repair should be recognized and treated with a pharyngoplasty. Extraocular muscle abnormalities are managed with eye patching, spectacles, and eye-muscle surgery, as appropriate. The recognition of cardiovascular and genitourinary malformations should lead to medical treatment and surgery. Cervical spine instability requires the decompression of any nerve impingement and the fusion of unstable segments. Unfortunately, the typical fixed neck curvature observed in patients with KFA cannot safely be straightened.
The aim of the reconstructive procedures used to correct a webbed neck is to create as normal a neck contour as possible with a symmetrical posterior hairline while avoiding obvious scarring.1 It is important to recognize that not all patients with webbed necks present with the same deformity. The clinician must assess the quality and quantity of available neck skin as well as the height and straightness of the cervical spine. Type I KFA consists of an extremely short skeletal neck that results in the increased width of the neck soft tissues. A spectrum of neck soft-tissue rearrangement procedures has been suggested to improve the webbed-neck deformity. Menick and colleagues modified the “butterfly” excision of redundant skin to avoid a midline scar.41 They did this by extending the more traditional inferior flaps into the scalp. Unfortunately, with this design, the scars are displaced laterally, and they become visible at the hairline, close to the original site of the web. Some surgeons feel that a midline posterior scar is often preferred aesthetically to one that can be seen along the lateral neck region. The placement of tissue expanders followed by flap rearrangements has also been carried out with some success. Thus, to review, procedures designed to correct the webbed neck suffer from an inability to elevate the hairline, noticeable scarring, and the intrinsically inadequate height of the skeletal structures of the neck.
The craniofacial skeletal deformities of patients with KFA typically present in one of two patterns.19,30,53 The first is in association with a congenital asymmetrical cervical spine fusion. In these patients, a variable degree of non-synostotic anterior plagiocephaly is present. The ipsilateral fronto–orbito–zygomatic flattening and retrusion may be significant enough that anterior cranial vault, orbital, and zygomatic reconstruction carried out through an intracranial approach will be indicated to adequately improve morphology (Fig. 31-1). These individuals will also have an asymmetrical dentofacial deformity that requires Le Fort I (maxillary) osteotomy, bilateral sagittal split ramus osteotomies of the mandible, and an osseous genioplasty procedure for three-dimensional repositioning (Fig. 31-2). The most unsettling aspect of the craniofacial reconstruction relates to the fact that the neck cannot be straightened. It will remain crooked, and so decisions about the best aesthetic positioning of the osteotomized craniofacial skeletal units must also be adjusted.
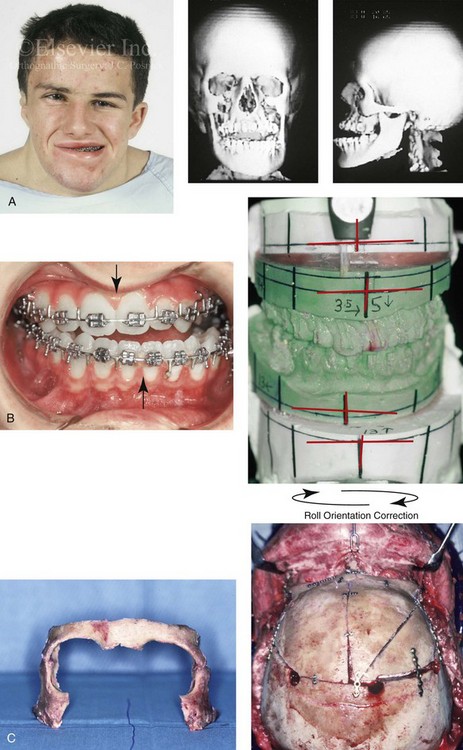
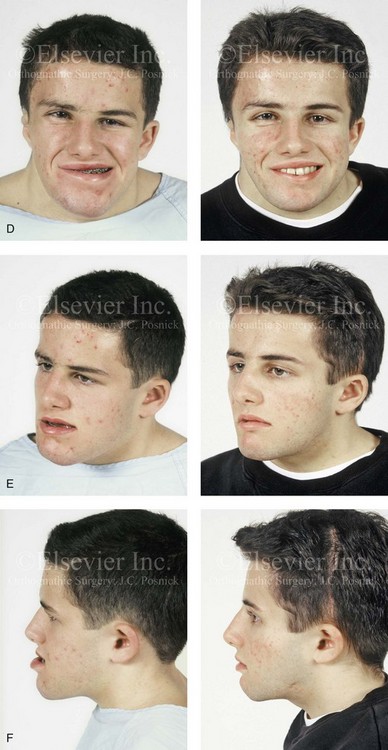
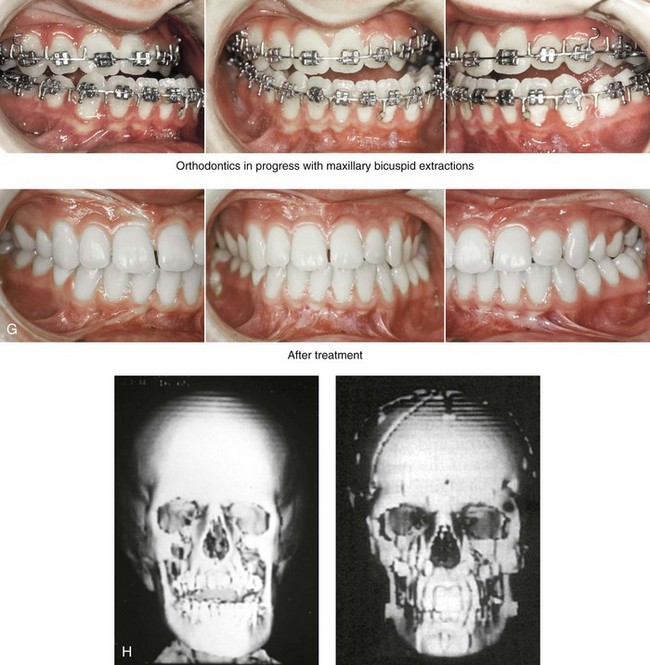
Figure 31-1 A 17-year-old boy with Klippel–Feil anomaly has a fixed head tilt as a result of a painless cervical spine fusion and a partial facial nerve palsy on the right side. There is marked asymmetry and deformity of the craniofacial region that involves the upper facial skeleton (i.e., the cranial vault, the orbits, and the zygomas) and the lower facial skeleton (i.e., the maxilla, the mandible, and the chin). The reconstruction was compromised by the fixed head tilt and the CN VII deficit. The patient underwent cranio–orbito–zygomatic (intracranial) reconstruction through a coronal scalp incision followed by orthognathic surgery that included a Le Fort I osteotomy, bilateral sagittal split ramus osteotomies, and an osseous genioplasty. A, Frontal facial and computed tomography scan views before surgery. B, Occlusal view before surgery and articulated dental cast that indicates analytic model planning. C, Intraoperative view of removed orbital and zygomatic units and intraoperative bird’s-eye view of the anterior cranial vault after cranio–orbito–zygomatic reconstruction. D, Frontal views with smile before and after reconstruction. Note that the fixed head tilt limited the the correction of facial symmetry. E, Oblique views before and after reconstruction. F, Profile views before and after reconstruction. G, Occlusal views with orthodontics in progress (including upper bicuspid extractions) and then after reconstruction. H, Three-dimensional computed tomography scan views before and after reconstruction.

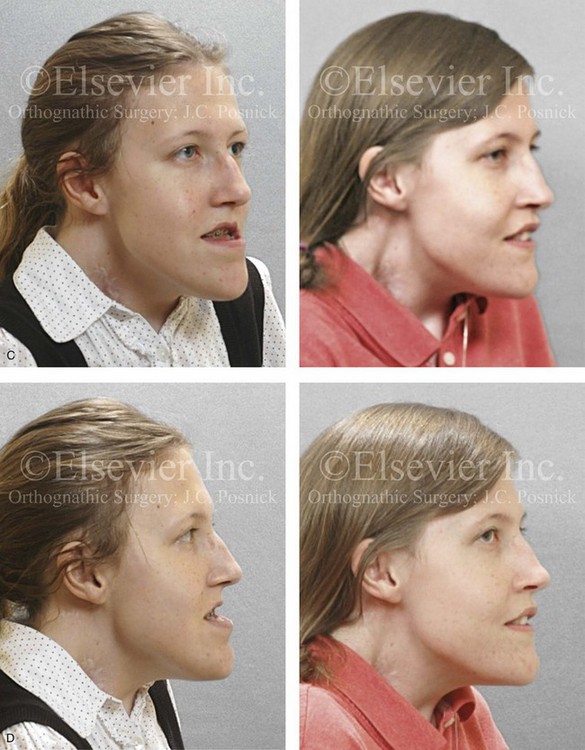
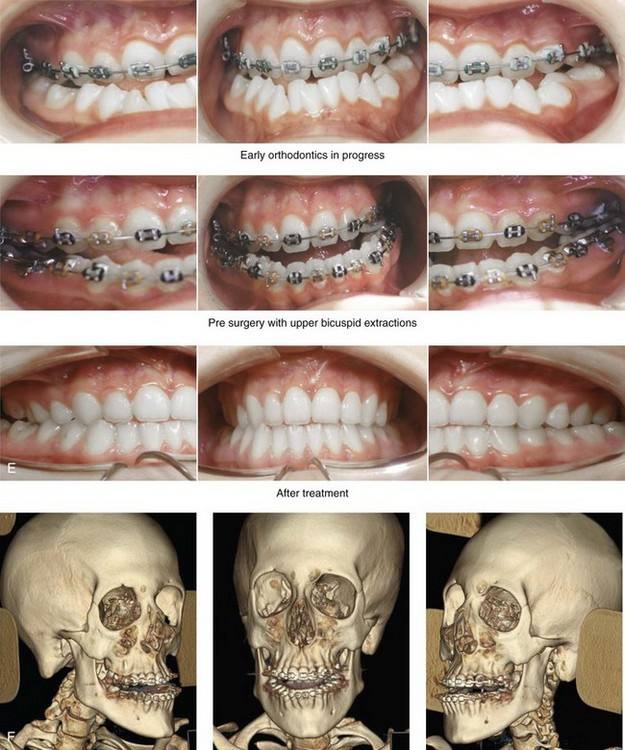

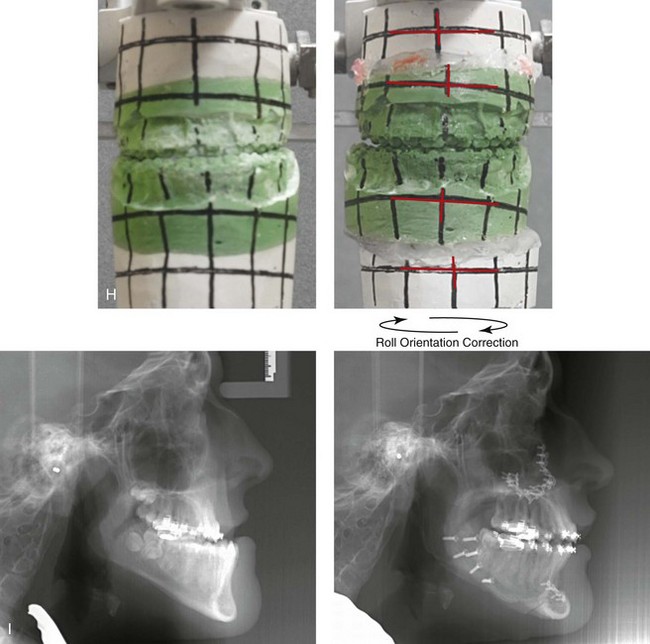
Figure 31-2 A teenage girl with the Klippel–Feil and Poland congenital anomalies has a fixed head tilt as a result of a painless cervical spine fusion. At birth, she was noted to have a significant diaphragmatic herniation of the stomach and spleen into the thoracic cavity. She underwent procedures for the correction of the diaphragmatic hernia and then required esophageal dilatations. She also required decompression and fusion for the cervical anomalies to limit injury to the upper spinal cord. She was referred to this surgeon as a teenager and underwent evaluations that included restorative dentistry, orthodontics, speech pathology, otolaryngology, pediatric surgery, pediatric orthopedic spine surgery, and pediatrics professionals. The assessment of pulmonary capacity, swallowing and dysphagia, and safe range of motion of the neck were of particular importance. There was marked asymmetry and deformity of the maxillomandibular region. The plans for reconstruction were affected by the fixed head tilt. The patient underwent a combined orthodontic and surgical approach, and her procedures included: maxillary Le Fort I osteotomy (horizontal advancement and cant correction); sagittal split ramus osteotomies (horizontal advancement and asymmetry improvement); osseous genioplasty (vertical shortening and horizontal advancement); and septoplasty and inferior turbinate reduction. Intraoperative cervical monitoring was carried out to limit the chance of spinal cord injury. A, Frontal views in repose before and after reconstruction. B, Frontal views with smile before and after reconstruction. C, Oblique facial views before and after reconstruction. D, Profile views before and after reconstruction. E, Occlusal views before treatment, with orthodontics in progress and with upper bicuspid extractions, and then after reconstruction. F, Computed tomography scan views before reconstruction. G and H, Articulated dental models that indicate analytic model planning. I, Cephalometric radiographs before and after reconstruction.
The second pattern of presentation is in association with a congenital symmetric cervical spine fusion. The cranio–orbito–zygomatic region is symmetrical and has relatively good proportions.53 No reconstruction procedures are generally required in the upper facial skeleton. With reference to the maxillomandibular region, the constant open mouth posture of the mandible generally results in vertical maxillary excess and a constricted maxillary arch width. The mandible is usually retrognathic, with a vertically long retrusive chin and an open-bite malocclusion. A degree of maxillomandibular asymmetry is expected. The neck–chin angle is markedly obtuse. These individuals will benefit from orthognathic surgery in combination with orthodontic treatment. The orthognathic surgery will generally require a Le Fort I osteotomy in segments to intrude the maxilla vertically, advance it horizontally, and widen it transversely. The preferred approach also involves bilateral sagittal split ramus osteotomies of the mandible with horizontal advancement and counterclockwise rotation to match the new maxillary position in combination with a vertical reduction and advancement genioplasty (see Chapter 15).
Down Syndrome (Trisomy 21 Syndrome)
Background
Speculation about the historic prevalence of Down syndrome has included references to depictions of the syndrome in 15th- and 16th-century paintings.69,81 Martínez-Frías described what seems to be the earliest observation of the syndrome in a sculpted terracotta head from 580 A.D.89 The terracotta piece comes from the Toltec culture of Mexico, and its facial features, which include macroglossia, help to define its subject as a person with Down syndrome. The features of this Down syndrome were initially described by Langdon Down in 1866, and the condition represents a combination of phenotypic features that includes a degree of mental deficiency and characteristic facial features.72
The birth prevalence of trisomy 21 syndrome is generally stated to be 1 in 650 live births, but it is known to vary in different populations from 1 in 600 to 1 in 2000 live births. During the 1970s, 15% of patients who had been institutionalized for mental deficiency were found to have trisomy 21 syndrome.75
About 95% of all cases of Down syndrome arise from non-disjunction. Approximately 80% are of maternal origin, with 20% being of paternal origin. The risk increases with increasing maternal age as well as with paternal age and particularly when the parents are more than 35 years old. It has been estimated that approximately 4.8% of Down syndrome cases arise from an unbalanced translocation, which may occur de novo or by parental transmission. Inherited D/G translocations occur in 93% of maternal cases and 7% of paternal cases; however, for G/G translocations, 50% are of maternal origin, and 50% are of paternal origin. It has been estimated that 65% to 80% of trisomy 21 conceptuses result in spontaneous abortions.75
Detectable mosaicism (found in about 3% of trisomy 21 cases) and the Down syndrome phenotype may only be partially expressed.75 The use of the DNA samples from individuals who have partial trisomy 21 with or without features of the phenotype has been helpful for understanding the nature of the disorder. The area between loci D21S58 and D21S42 has been identified as being associated with mental retardation and with most of the facial features of Down syndrome. The facial features that have been identified in this region include oblique palpebral fissures; epicanthal folds; a flat nasal bridge; a protruding tongue; short, broad hands with clinodactyly of the fifth finger; a gap between first and second toes; hypotonia; short stature; Brushfield spots; and characteristic dermatoglyphic findings, including a single palmar crease and an increased number of ulnar loops.88
Individuals with Down syndrome have a specific set of major congenital malformations, including heart anomalies (30% to 40%) and gastrointestinal tract anomalies such as duodenal stenosis or atresia, imperforate anus, and Hirschsprung disease.75 The development of Alzheimer disease occurs at a much earlier age in individuals with trisomy 21 than it does in the general population. The frequency of symptomatic gallbladder disease is 25% higher among adults with Down syndrome as compared with the general population.
Down syndrome is characterized by extensive phenotypic variability.65,67,71,98 Although cognitive impairment, muscle hypotonia at birth, and dysmorphic features occur to some extent in all individuals, most associated traits occur in only a fraction of affected individuals. In the patient with Down syndrome, a number of characteristic facial features that vary from patient to patient are frequently recognized:
2. Oblique (anti-mongoloid) slanting of the eyelids
3. Strabismus as a result of extraocular muscle dysfunction
4. Flatness of the dorsum of the nose
5. Hypoplasia of the upper jaw that results in an Angle Class III malocclusion
In 1967, Otermin Aguirre presented a series of patients with Down syndrome who underwent reconstructive surgery in an attempt to normalize their facial morphology.93 During the same year, Hohler attempted to normalize the face of a young girl with Down syndrome by augmenting the hypoplastic dorsum of the nose and the receding chin with alloplastic implants.77 In 1977, Lemperle and Spitalny systematically tried to normalize the faces of children with Down syndrome.82–85 Similar results were published by Olbrisch,91,92 Patterson and colleagues,94 Wexler and Peled,103,104 and Rozner,99 in addition to several others.76,90,94,97
Despite the initial enthusiasm for facial reconstruction in children with Down syndrome during the late 1960s through the mid 1980s, a critical review of the procedures used to normalize the face was not carried out until the mid to late 1980s. In 1989, Katz and Kravetz presented a balanced, thoughtful review and a discussion of the literature regarding the “effectiveness of facial surgery for persons with Down syndrome.”79 The research of Katz and Kravetz may be summarized as follows: If improved functioning, appearance, and social acceptability are measured by parent and treating physician satisfaction, then the outcome of facial plastic surgery for people with Down syndrome seems positive. If function, appearance, and social acceptability are evaluated by people who were not involved in making the decision to conduct the facial plastic surgery—and particularly when control subjects are included in the evaluation—then the outcome of facial plastic surgery for people with Down syndrome is not so positive. For most patients with Down syndrome, the surgeons’ and parents’ well-intentioned attempts to normalize the child’s facial morphology could not be realized.96
Macroglossia with protrusion of the large tongue is a frequent feature of Down syndrome. Dystonia or hypotonia of the tongue is also a factor.66,74 During the 1970s and the early 1980s, several investigators suggested that partial glossectomy in the patient with Down syndrome could improve the clarity of speech.68,70 However, studies that used objective criteria to document speech results after partial glossectomy in patients with Down syndrome showed no significant differences in the number of articulation errors between audiotape recordings made preoperatively and 6 months postoperatively in a series of patients or between the recordings of the same patients and recordings of an additional series of Down syndrome control patients who did not undergo surgery. A further study by Margar-Bacal and colleagues looked at changes in the aesthetic appearance and intelligibility of speech after partial glossectomy in patients with Down syndrome.87 The aesthetic appearance of speech (i.e., the visual acceptability of the patient while he or she is speaking) was judged from visual information only. Judgments of speech intelligibility were made separately from the auditory portion of the videotapes. The acceptability and intelligibility of the children were also judged together during audiovisual presentation. Statistical analysis showed that speech was significantly more acceptable aesthetically after surgery. No significant difference was found in speech intelligibility postoperatively as compared with preoperatively.
An Angle Class III malocclusion as a result of the midface hypoplasia with overclosure of the mandible is a frequent finding in patients with Down syndrome.95 This was objectively studied by Farkas with the use of anthropometric measurements in which he documented that 60% of children with Down syndrome showed a disproportionately small depth to the middle third of the face (i.e., the anthropometric measurement from the subnasal point to the tragus).73 The congenital absence of permanent teeth, poor root formation, and hypoplasia of the enamel are frequent complicating factors.
Author’s Approach to Facial Reconstruction in Individual’s with Down Syndrome
When a child with Down syndrome approaches skeletal maturity (i.e., 14 to 16 years of age for girls, 16 to 18 years of age for boys) and a significant jaw deformity with malocclusion is present, then a comprehensive approach of orthodontic treatment and orthognathic surgery can effectively alter the facial proportions (aesthetics) and improve head and neck functions (i.e., lip posture, chewing, speech articulation, and breathing) (Fig. 31-3). A formal sleep study is indicated before surgery to clarify the extent of upper airway obstruction and the degree of maxillomandibular advancement required to manage it.1,78,80,86,100–102 The orthodontic treatment may be compromised as a result of poor dental root formation, congenitally missing teeth, or limited compliance. The coordination of care with an appropriate dental team (e.g., a restorative dentist, a periodontist, an orthodontist) is useful before the initiation of any treatment.
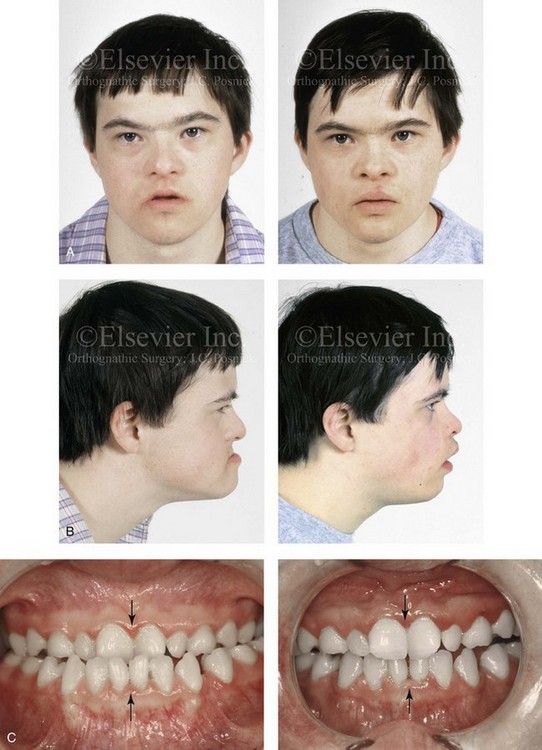
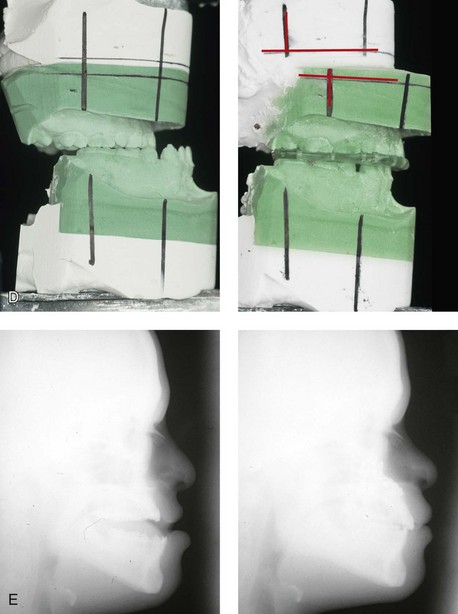
Figure 31-3 A 16-year-old boy with Down syndrome presented with a jaw deformity and malocclusion. He underwent a combined orthodontic and surgical approach. The surgery was limited to a Le Fort I osteotomy (horizontal advancement and vertical lengthening). A, Frontal views before and after reconstruction. B, Profile views before and after reconstruction. C, Occlusal views before and after reconstruction. Note that the orthodontic and restorative dental objectives were limited by congenital malformations of the dental crowns and dental roots. D, Articulated dental casts that indicate analytic model planning. E, Lateral cephalometric radiographs before and after reconstruction.
The correction of the dentofacial deformity and the management of the airway frequently requires maxillary Le Fort I osteotomy (horizontal advancement and vertical lengthening), often with sagittal split ramus osteotomies for alignment to the new maxillary position (i.e., advancement but not setback) and an osseous genioplasty (horizontal advancement and vertical lengthening). Septoplasty and inferior turbinate reduction should be simultaneously performed if required to improve nasal airflow (see Chapter 10).
Neurofibromatosis
Background
In 1849, Robert Smith, a professor of surgery at Dublin Medical School, reported clinical and necropsy findings in two cases of individuals who were presumed to have neurofibromatosis; he cited 75 references to similar patients from the earlier medical literature.130 Unfortunately, he did not recognize that the tumors (neurofibromas) contained neural elements. In 1882, von Recklinghausen published his findings, which convinced the medical world that neurofibromatosis was a distinct entity of neural origin.165 Today, neurofibromatosis is known to be etiologically heterogeneous. Cohen and Hayden were the first to differentiate Proteus syndrome from neurofibromatosis in 1979.119,162 Types I and II are the forms that are frequently encountered by the craniomaxillofacial surgeon.
Neurofibromatosis Type II (Acoustic Type)
The hallmark of Type II neurofibromatosis is the presence of bilateral acoustic neuromas.147,150 Symptoms are usually caused by pressure on the vestibulocochlear and facial nerve complex. The first symptom is usually hearing loss that often begins during the teenage years or the early 20s. Occasionally, hearing loss may occur as early as the first decade or as late as the seventh decade of life. Cafe-au-lait spots and cutaneous neurofibromas are also present, but they are seen less frequently than they are among patients with neurofibromatosis Type I.121 Tumors of the central nervous system are especially common, with Schwann cell tumors occurring most frequently. Multiple tumors of meningeal or glial origin may also occur.147 Neurofibromatosis Type II has autosomal dominant inheritance with penetrance of more than 95%. The responsible gene has been mapped to chromosome 22.
Neurofibromatosis Type I (von Recklinghausen Type)
Joseph Merrick, the Elephant Man, has often been thought to have had von Recklinghausen disease. However, after considering several diagnostic possibilities, Cohen concluded that Merrick’s skeletal findings are most consistent with Proteus syndrome.119,130,162
This classic form of neurofibromatosis accounts for approximately 90% of all affected cases. Major features include six or more cafe-au-lait spots, cutaneous neurofibromas, and Lisch nodules.130 Axillary freckling develops in about 66% of all patients. Neurofibromatosis type I, which is the classic form of neurofibromatosis, is the one described by von Recklinghausen; it is also the most frequent form, occurring in 1 of every 2500 to 3000 births.146,160,165,166 Inheritance is autosomal dominant, with almost 100% penetrance. Approximately 50% of cases represent new mutations, and an increase in paternal age at the time of conception has been found to be an associated feature. Although most evidence points to neurofibromatosis as a disorder of neural crest derivation, some controversy remains about whether the neural and mesenchymal components are interrelated or if they may arise independent of each other.110,116,153
Type I is caused by a mutation of the neurofibromin 1 (NF1) gene. Approximately 5% to 20% of all patients with Type I neurofibromatosis patients carry a heterozygous deletion of approximately 1.5 Mb that involves the NF1 gene and the contiguous change line in its flanking region.157 Miller and Hall found that patients born to affected mothers have more severe disease than those born to affected fathers.152 The mutation rate in the NF1 gene is one of the highest known in humans, with approximately 50% of all patients with NF1 presenting with novel mutations.124
The natural history of neurofibromatosis Type I has been reviewed by several authors.138 More than 40% of patients have some clinical manifestations at birth, and more than 60% have manifestations by the second year of life. Cafe-au-lait spots usually develop first, with multiple lesions present within the first year of life. In approximately 50% of patients, axillary freckling appears later. Cutaneous neurofibromas appear around the onset of puberty and increase in number throughout life.138 Lisch nodules begin to appear during early childhood and have been observed in almost all affected adults. Plexiform neurofibromas occur in 30% of these patients.
Approximately 33% of all patients develop one or more complications in association with the disease.105–108,115 It has been estimated that some form of malignancy develops in 6% of patients with neurofibromatosis type I who are more than 18 years old.109,117,118 Other important complications that may occur are neurologic and include epilepsy, aqueduct stenosis, and spinal neurofibromas. Learning disabilities of various kinds affect 25% of patients.137,154,163
Any part of the eye may be involved.111,126,128,132 Lisch nodules of various sizes may be found anywhere in the iris. These lesions are melanocytic hamartomas, and they are found only in patients with neurofibromatosis. Phakoma, congenital glaucoma, corneal opacity, detached retina, optic atrophy, and congenital ptosis of the eyelids have all been reported. Intraorbital lesions may also produce proptosis and eye-muscle palsies.
The literature concerning the presenting head and neck dysmorphology and the suggested treatment of orbitotemporal neurofibromatosis is extensive.* The plexiform neurofibroma that involves the orbit, the eyelid, and the temporal region may also have an intracranial component. It generally appears during childhood as a swelling of the upper eyelid, and it slowly and progressively worsens (Fig. 31-4). Further involvement of the subcutaneous tissues of the eyelids with varying degrees of ptosis and proptosis of the globe also occurs. For patients in whom progression continues, there is further plexiform neurofibromatosis of the temporal area, pulsating proptosis, continued enlargement of the eyelids with extensive mechanical ptosis, and the inability to open the eye, which results in severely diminished visual acuity or blindness. The globe itself is involved with neurofibroma, and buphthalmos may be present. The eye may be painful as a result of infection and epiphora.
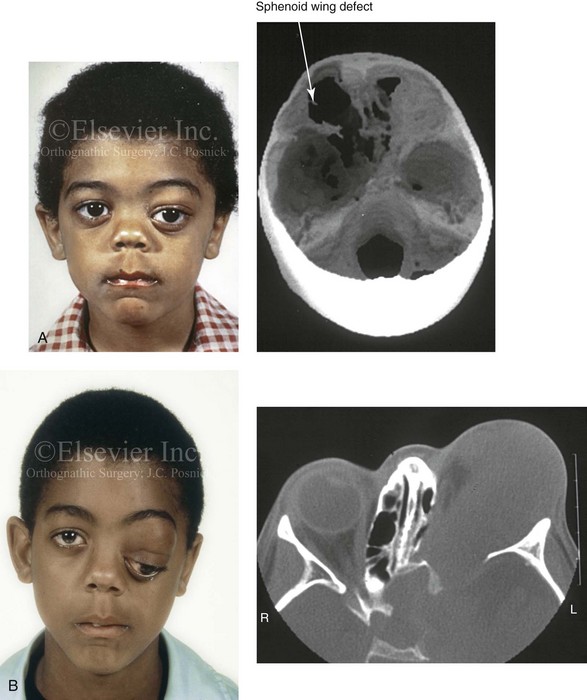
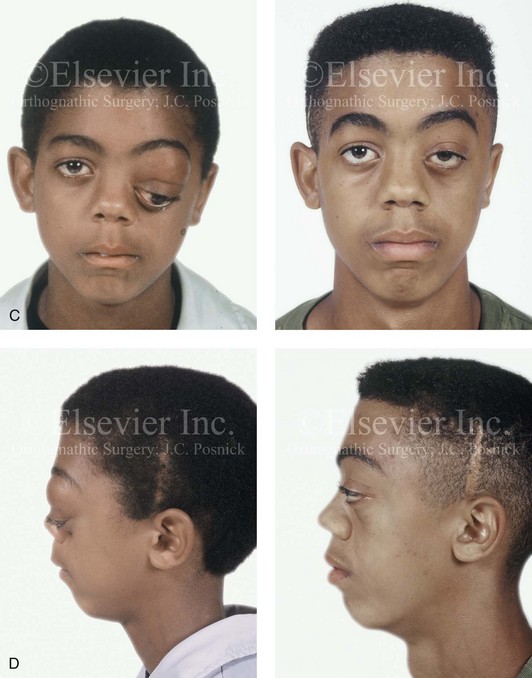
Figure 31-4 A child with neurofibromatosis involving the left cranio–orbital region. Aplasia of the sphenoid wing and the gradual growth of plexiform neurofibroma occurred, which extended from the cavernous sinus through the widened superior orbital fissure, through the cone of the eye, and into the upper eyelid. Despite an intracranial debulking procedure carried out by another surgeon during the patient’s middle childhood years, the boy presented to this surgeon with residual and recurrent plexiform neurofibroma that resulted in the distortion and proptosis of the left eye. Vision remained in the affected eye, and the decision was made to preserve it. The patient underwent intracranial reconstruction of the cranio–orbito–zygomatic skeleton, including the debulking of the soft-tissue tumor and the vertical and horizontal debulking of the upper eyelid. Intracranial debulking with eye presentation was accomplished. A, Frontal view during early childhood and a computed tomography scan of the anterior skull base indicating the defect in the sphenoid wing. B, Frontal view at 10 years of age. Despite a debulking procedure carried out by another surgeon, residual growth has reoccurred. An axial computed tomography scan view through the midorbits indicates the extent of plexiform neurofibromatosis from the cavernous sinus through the upper eyelid. C, Frontal views before and after reconstruction. Note that the surgical correction of the upper eyelid ptosis was simultaneously carried out but it was limited to maintain adequate corneal protection. D, Profile views before and after reconstruction.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


