Syndromes with Craniosynostosis
Evaluation and Treatment
• Mutations in Craniosynostosis and Craniosynostosis Syndromes
• Neurologic Aspects of Craniosynostosis
• Effects of Midface Deficiency on the Upper Airway
• Surgical Approach to Craniosynostosis Sydromes
• Staging of Reconstruction for Crouzon Syndrome
General Considerations
Craniosynostosis, which is the premature fusion of cranial sutures, affects approximately 1 in 2500 children. Patients may present with a wide range of phenotypic and functional deformities.94 Virchow’s law from 1851 states that the premature fusion of a cranial vault suture results in growth arrest perpendicular to the affected suture and compensatory growth parallel to the fused suture.16,164,269 This principle is generally applicable to single sutures; multisutural synostosis—such as the combination of sagittal and lambdoid synostosis (coup de sabre) or a cloverleaf skull—can assume more complex patterns of cranial morphology.43,46,47 In addition to the type of affected suture, the eventual head shape depends on the timing and order of cranial suture fusion. Craniosynostosis may be of prenatal or perinatal onset, or it may occur during infancy.14,74 The earlier synostosis occurs, the more dramatic the effect on subsequent cranial growth and development; the later synostosis occurs, the less effect on cranial growth and development.44
Active pediatric neurosurgical services generally see a characteristic frequency, gender predilection, and phenotypic presentation for sutural involvement.6,15,55,125,133,154,161,193,232,238,243,278,283 Sagittal synostosis occurs most commonly, with a higher incidence among boys than girls. Patients typically present with a boat-shaped skull (i.e., scaphocephaly) in combination with midline ridging in the posterior half of the skull. Metopic synostosis is currently the second most frequent form of synostosis. Presentations occur along a spectrum that ranges from metopic ridging to the presence of a triangular-shaped skull (i.e., trigonocephaly). It should be noted that metopic ridging is common during infancy and childhood and that it is not necessarily associated with a diagnosis of craniosynostosis. Coronal synostosis occurs next most frequently; girls slightly predominate or, in some surveys, boys and girls have equivalent frequency.46 Unicoronal synostosis is marked by frontal plagiocephaly, whereas bicoronal synostosis is characterized by a short and wide skull (i.e., brachycephaly). Multiple synostoses of various types are less common than coronal synostosis. Lambdoid synostosis occurs with least frequency, and it is marked by occipital plagiocephaly.44
Craniosynostosis is etiologically heterogeneous and pathogenetically variable. Overall, 8% of cases have a familial component, and this is generally marked by an autosomal dominant inheritance pattern. Specifically, pedigrees are familial in 14.4%, 6%, and 5.6% of coronal, sagittal, and metopic synostoses, respectively.44 Although some pedigrees show synostosis of a specific suture, others may show fusion of different sutures in affected relatives of the same family. Different chromosomal aberrations have been linked to craniosynostosis, with variable penetrance. In some conditions the penetrance is high, such as with dup(3q), del(7p), del(9p), and del(11q). In other cases, penetrance can be very low, such as with dup(5p), del(6)(22q22.2-q23.1), del(8q), and dup(15q).44
Many syndromes are associated with craniosynostosis, and well over 100 are known.44 It is important to accurately diagnose syndromal patients for three reasons.22 First, most syndromes with craniosynostosis affect not only the cranial vault but also the cranial base and the midface. Deficiencies in these skeletal sites, which are variable in degree yet commonly serious, must be addressed as part of the staged reconstructive approach. Second, syndromes are often genetic and familial, thus necessitating proper counseling. Third, depending on the experience of the particular surgeon, the molecular diagnosis should be made. Often, the patient may be referred for molecular diagnosis and counseling to avoid the possibility of litigation.37,43,46,47
Primary forms of craniosynostosis are most common and include single- and multi-sutural synostosis. Some conditions, however, result in secondary synostosis. These include hyperthyroidism; rickets; mucopolysaccharidoses, such as Hurler syndrome and Morquio syndrome; hematologic disorders, such as thalassemia and sickle cell anemia; teratogens, such as diphenylhydantoin, retinoids, valproate, and aminopterin; and certain malformations, such as holoprosencephaly and microcephaly.44
Mutations in Craniosynostosis and Craniosynostosis Syndromes
Mutations responsible for craniosynostosis have been identified in FGFR1, FGFR2, FGFR3, TWIST, MSX2, EFNB1, RAB23, EFNA4, POR, and ALPL.44 These include Apert syndrome (two common FGFR2 mutations; Ser252Trp, Pro253Arg), Crouzon syndrome (more than 30 FGFR2 mutations), Pfeiffer syndrome (more than 30 FGFR2 mutations), are known and at least 6 of them are the same as those found with Crouzon syndrome; one Pfeiffer syndrome (mutation on FGFR1).* In addition, there are some cases of FGFR2 mutations with isolated coronal synostosis as well as with some other forms of non-syndromic synostosis. A single known FGFR2 mutation is known for Jackson-Weiss syndrome.1,41,182 Although this disorder is distinctly uncommon, affected families are often large, with variable phenotypic expression. Beare-Stevenson cutis gyrate syndrome involves one of two possible mutations in the transmembrane domain of FGFR2.37,42
Muenke syndrome is the most common craniosynostosis syndrome that is currently known. It involves a single mutation on FGFR3, and it is characterized most commonly by unilateral coronal synostosis and less commonly by bicoronal synostosis. About 6% of affected patients have macrocephaly without synostosis, and cloverleaf skull has been reported in some instances.37,42 Therefore, all patients with coronal synostosis should be tested for this very common FGFR3 mutation; if they are negative, they should be tested for FGFR2.
Crouzonodermoskeletal syndrome is characterized by one specific FGFR3 mutation. The name indicates its characteristic features: either a Crouzonoid or cloverleaf skull appearance; acanthosis nigricans; and a decreased interpediculate distance that is radiographically present but not clinically significant. Serious decreased interpediculate distance is a feature of the most common mutation for achondroplasia, which is only 11 amino acids away from the FGFR3 mutation for Crouzonodermoskeletal syndrome. To call this disorder “Crouzon syndrome with acanthosis” is unwarranted, because only one FGFR3 mutation is responsible for all cases; this is in contrast with Crouzon syndrome, which involves more than 30 different FGFR2 mutations.38
Rarely, patients with achondroplasia and hypochondroplasia (both of which involve mutations on FGFR3) may also have craniosynostosis (there have been three reported cases of hypochondroplasia and two reported cases of achondroplasia). Saethre–Chotzen syndrome has TWIST mutations inside or outside of the coding region that result in haploinsufficiency.48 Craniofrontonasal syndrome is caused by heterozygous loss-of-function mutations in EFNB1. EFNA4 mutations rarely cause non-syndromal coronal synostosis. Carpenter syndrome, which is an autosomal recessive disorder, is caused by RAB23 mutations. Boston-type craniosynostosis, which is an autosomal dominant disorder caused by MSX2 mutations, presents with variable expressivity (coronal synostosis, frontal depression, or cloverleaf skull). Antley–Bixler syndrome is caused by POR mutations, and infantile hypophosphatasia is caused by ALPL mutation.44,184
Complex Craniosynostosis
Background
Complex craniosynostosis, which involves the fusion of multiple cranial sutures, occurs in about 5% of non-syndromic cases.47 Two thirds of cases involve two affected sutures, whereas one third of cases involve more than two affected sutures.203 As the number of affected sutures increases, so does the risk of intracranial hypertension and associated mental deficiency. As demonstrated by Renier and colleagues, the incidence of intracranial hypertension rises from 14% to 47% when going from single to several affected sutures.228,226,227,229,230 In addition, increasing sutural involvement can be accompanied by worsened phenotypic severity. In a series reported by Chumas and colleagues,34 59% of cases of sagittal and bilateral lambdoid synostosis had an acute angulation of the posterior skull, which is known in French as coup de serpe and means “cut with a scythe.” In a similar fashion, marked anterior dysmorphism with frontal bone hypoplasia was found in 23% of cases of metopic and bilateral coronal synostosis.
Cloverleaf skulls, which represent the extremes of phenotypic severity, are pathogenetically variable (Fig. 30-1).30,204 Synostosis may involve the coronal, lambdoid, and metopic sutures, which are marked by bulging of the cerebrum through an open sagittal suture or, in some cases, through patent squamosal sutures. Synostosis of the sagittal and squamosal sutures with cerebral eventration through a widely patent anterior fontanel may also be observed. A trilobular skull shape may also occur, with complete synostosis of all cranial sutures in some cases or with widely patent sutures and no craniosynostosis at birth in other instances.36,51 In addition to being pathogenetically variable, cloverleaf skulls are also etiologically heterogeneous. This condition most commonly occurs with type 2 thanatophoric dysplasia (i.e., in about 40% of cases) with a specific FGFR3 mutation, but stillborn status or an early demise during very early infancy precludes treatment. Isolated cloverleaf skull occurs in about 20% of cases. It also occurs in about 15% of cases of type 3 Pfeiffer syndrome, and, although the prognosis is guarded, these patients can be treated aggressively.45,48
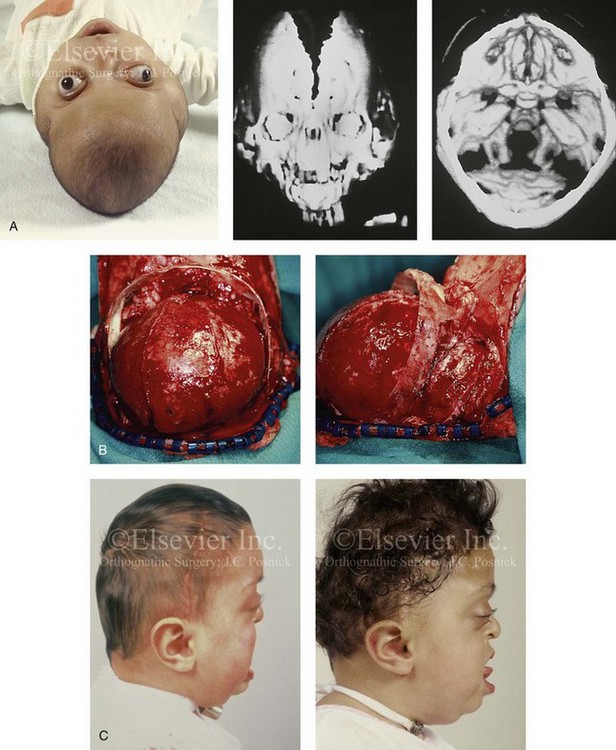
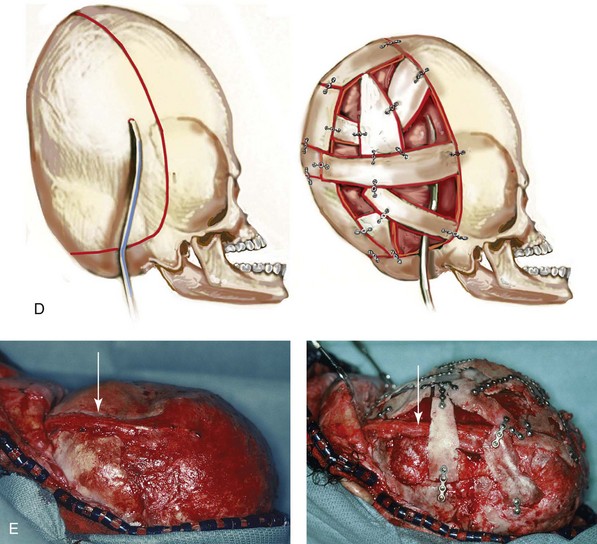
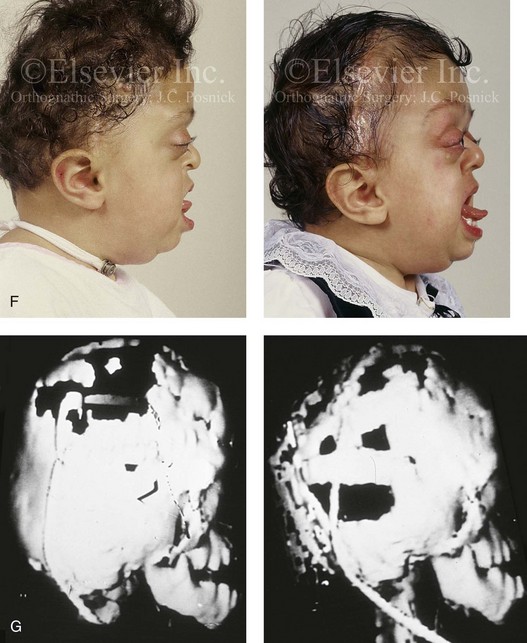
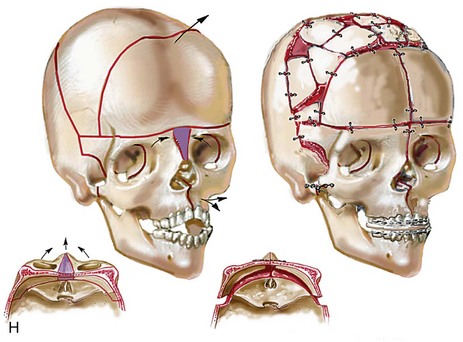
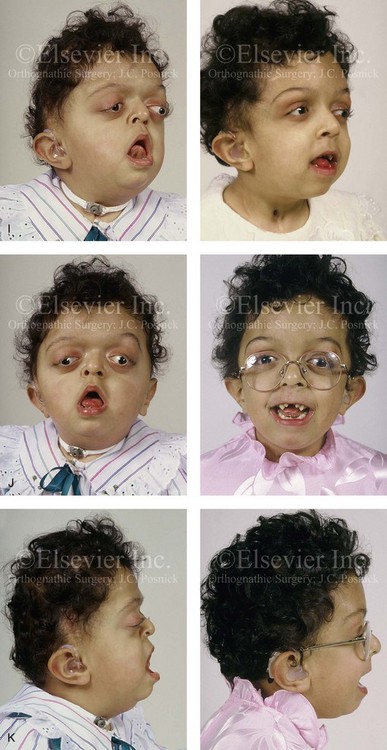
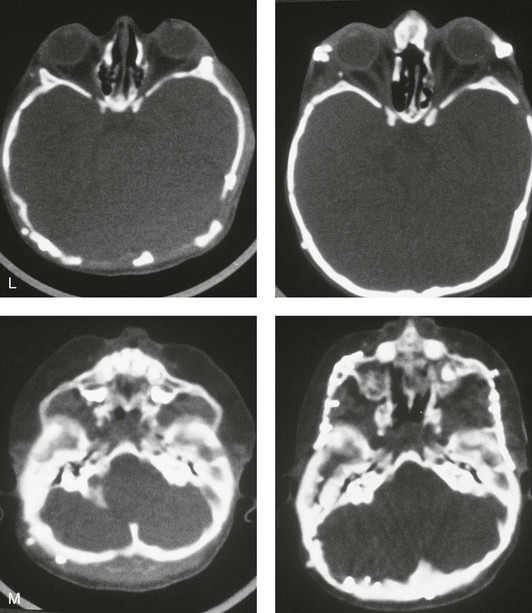
Figure 30-1 A child with a severe form of cloverleaf skull anomaly. At 10 months of age, she was referred to this surgeon and underwent first-stage cranial vault and upper orbital decompression with reshaping. She then required a ventriculoperitoneal shunt for the management of hydrocephalus. When the patient was 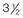 years old, posterior cranial vault decompression with reshaping to increase the intracranial volume was performed. When she was
years old, posterior cranial vault decompression with reshaping to increase the intracranial volume was performed. When she was 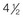 years old, facial bipartition osteotomies in combination with anterior cranial vault reshaping and advancement were carried out. After the cranial vault and facial bipartition procedures, the patient’s airway improved, and it was possible to remove the tracheostomy tube. The cranial vault reshaping expanded the intracranial volume, thereby providing more space for the brain. The midface advancement improved the eye proptosis as well as the patient’s ability to chew and articulate speech. As part of the staged reconstruction, she will require orthognathic surgery in combination with orthodontic treatment at the time of early skeletal maturity. A, Frontal facial and computed tomography (CT) scan views at 10 months of age. B, Intraoperative views at 10 months of age after craniotomy and the removal of the cranial vault and the upper orbits. The “bandeau” is in place (3 cm advancement) before the reconstruction of the overlying cranial vault. C, Profile views at
years old, facial bipartition osteotomies in combination with anterior cranial vault reshaping and advancement were carried out. After the cranial vault and facial bipartition procedures, the patient’s airway improved, and it was possible to remove the tracheostomy tube. The cranial vault reshaping expanded the intracranial volume, thereby providing more space for the brain. The midface advancement improved the eye proptosis as well as the patient’s ability to chew and articulate speech. As part of the staged reconstruction, she will require orthognathic surgery in combination with orthodontic treatment at the time of early skeletal maturity. A, Frontal facial and computed tomography (CT) scan views at 10 months of age. B, Intraoperative views at 10 months of age after craniotomy and the removal of the cranial vault and the upper orbits. The “bandeau” is in place (3 cm advancement) before the reconstruction of the overlying cranial vault. C, Profile views at 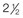 and then at
and then at 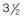 years of age with flattened posterior cranial vault and severe midface deficiency. D, Illustration of flattened posterior cranial vault with the ventriculoperitoneal shunt in place. Illustration after posterior cranial vault reshaping is also shown. E, Intraoperative side view with the patient in the prone position before and after the craniotomy and the reshaping of the posterior cranial vault. Arrows point to the ventriculoperitoneal shunt, which remains intact and deep to the skull reconstruction. F, Profile view at
years of age with flattened posterior cranial vault and severe midface deficiency. D, Illustration of flattened posterior cranial vault with the ventriculoperitoneal shunt in place. Illustration after posterior cranial vault reshaping is also shown. E, Intraoperative side view with the patient in the prone position before and after the craniotomy and the reshaping of the posterior cranial vault. Arrows point to the ventriculoperitoneal shunt, which remains intact and deep to the skull reconstruction. F, Profile view at 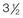 years of age, before and after posterior cranial vault reconstruction. G, CT scan views before and just after posterior cranial vault reshaping. H, Illustration before facial bipartition. The second illustration indicates the planned reconstruction. I, Oblique facial views before and after facial bipartition reconstruction. The tracheostomy has been removed. J, Facial views before and after facial bipartition reconstruction. K, Profile views before and after facial bipartition reconstruction. L, Axial CT views through the midorbits before and after reconstruction. M, Axial CT views through the zygomatic arches before and after reconstruction. A, B, C (right), D, E, F, H (left), I, K, L, M, From Posnick JC: The craniofacial dysostosis syndromes: secondary management of craniofacial disorders, Clin Plast Surg 24:429-446, 1997.
years of age, before and after posterior cranial vault reconstruction. G, CT scan views before and just after posterior cranial vault reshaping. H, Illustration before facial bipartition. The second illustration indicates the planned reconstruction. I, Oblique facial views before and after facial bipartition reconstruction. The tracheostomy has been removed. J, Facial views before and after facial bipartition reconstruction. K, Profile views before and after facial bipartition reconstruction. L, Axial CT views through the midorbits before and after reconstruction. M, Axial CT views through the zygomatic arches before and after reconstruction. A, B, C (right), D, E, F, H (left), I, K, L, M, From Posnick JC: The craniofacial dysostosis syndromes: secondary management of craniofacial disorders, Clin Plast Surg 24:429-446, 1997.
Plagiocephaly is defined as the asymmetric distortion of the skull. Well-known types include synostotic anterior plagiocephaly (unilateral coronal synostosis), synostotic posterior plagiocephaly (unilateral lambdoid synostosis), deformational anterior plagiocephaly, and deformational posterior plagiocephaly.146,148,268 Among these types, deformational posterior plagiocephaly is common, whereas unilateral lambdoid synostosis is rare. Deformational posterior plagiocephaly can be caused by intrauterine factors such as hypotonia, fetal positioning, and prematurity. This can produce asymmetric flattening of the occiput that becomes favored by infants sleeping on their backs, thereby exaggerating the plagiocephaly. Deformational posterior plagiocephaly has also increased dramatically since the 1992 “Back to Sleep” campaign by the American Academy of Pediatrics, which calls for supine infant sleeping to reduce the risk of sudden infant death syndrome.44 Finally, rare forms of plagiocephaly can be produced by unilateral frontosphenoidal synostosis and unilateral frontozygomatic synostosis.44
Apert Syndrome
Apert syndrome is characterized by craniosynostosis, midface deficiency, symmetric syndactyly of the hands and feet, and other abnormalities* (Figs. 30-2 through 30-5). Most cases are associated with brachycephaly secondary to bicoronal synostosis. Megalencephaly and increased cranial height are characteristic of Apert syndrome; affected children typically display head circumference, length, and weight that are above the normal 50th percentile. Benign distortion ventriculomegaly is also a feature of Apert syndrome, and patent sutures and synchondroses (except for coronal synostosis) are present as well. The frequency of progressive hydrocephalus is much lower than in it is with Crouzon and Pfeiffer syndromes. Central nervous system anomalies may include hypoplasia of the corpus callosum, agenesis of the corpus callosum, agenesis of the septum pellucidum, cavum septum pellucidum, dorsally displaced hippocampi and hippocampal gyri, hypoplastic white matter, and heterotopic gray matter.46,48
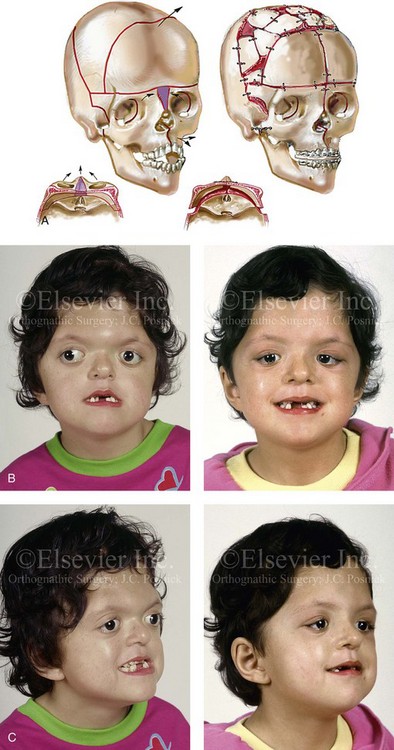
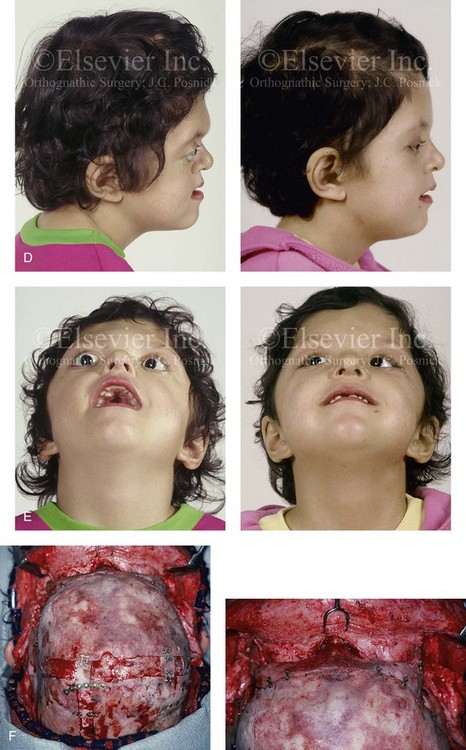
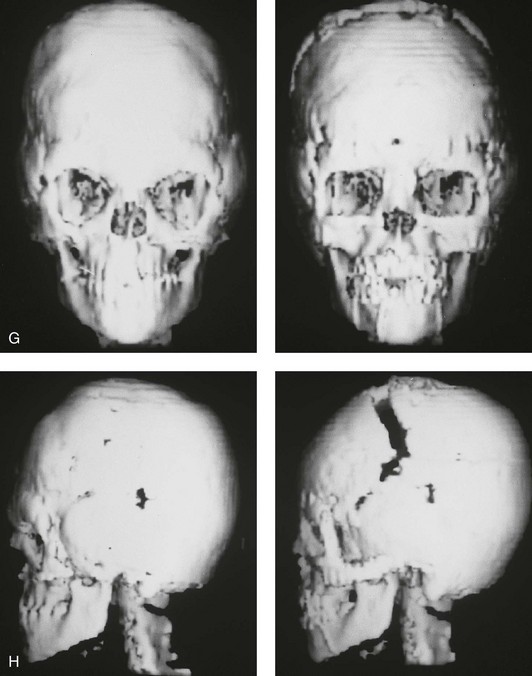
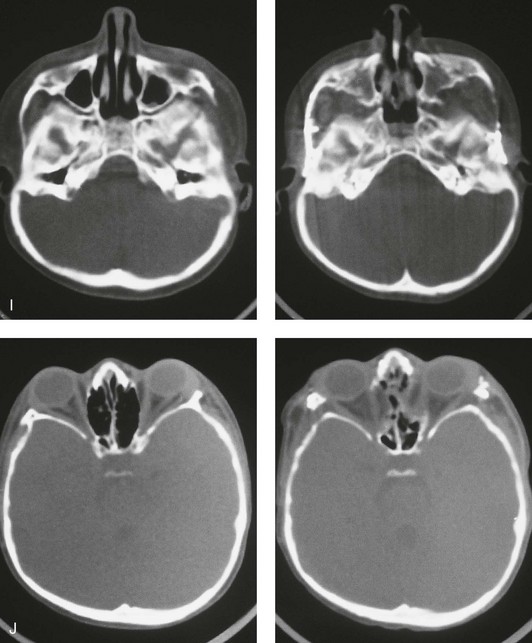
Figure 30-2 A child who was born with Apert syndrome underwent a bilateral lateral canthal advancement procedure when she was 6 weeks old; this was carried out by a neurosurgeon working independently. When she was 18 months old, she returned with turricephaly and a constricted anterior cranial vault that required further cranio–orbital decompression and reshaping. When she was 5 years old, she underwent anterior cranial vault and facial bipartition osteotomies with reshaping. As part of the staged reconstruction, she will require orthognathic surgery and orthodontic treatment during her teenage years. A, Illustrations of craniofacial morphology before and after the facial bipartition osteotomies and reconstruction. B, Frontal views before and after facial bipartition reconstruction. C, Oblique facial views before and after facial bipartition reconstruction. D, Profile views before and after facial bipartition reconstruction. E, Worm’s-eye views before and after facial bipartition reconstruction. F, Intraoperative views after facial bipartition and anterior cranial vault reconstruction. G and H, Computed tomography scan views before and early after reconstruction. I, Axial computed tomography scan views through the zygomas before and after reconstruction. J, Axial computed tomography scan views through the midorbits before and after reconstruction indicating relief of proptosis.
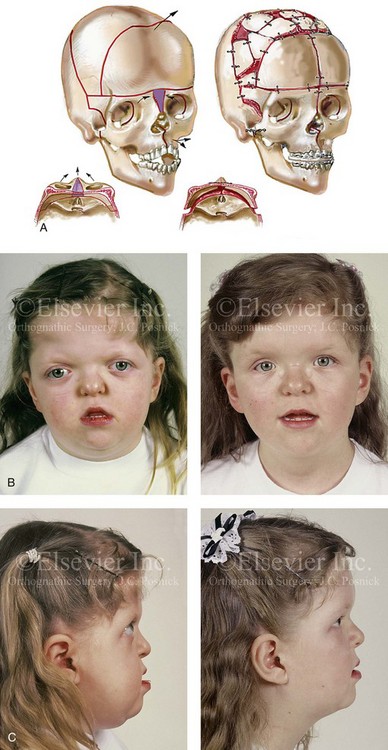
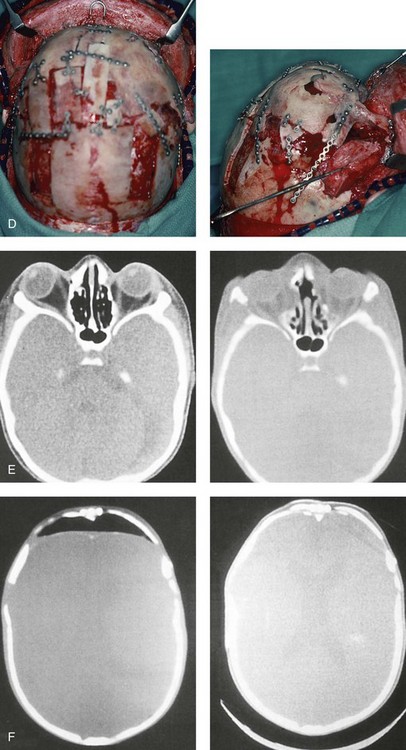
Figure 30-3 A 5-year-old girl with Apert syndrome who underwent lateral canthal advancements when she was 6 months old and in the care of another surgeon. She then presented to this surgeon with residual craniofacial deformities that required anterior cranial vault and facial bipartition osteotomies with reshaping. She will require orthognathic surgery and orthodontic treatment during her teenage years to complete the reconstruction. A, Illustrations of preoperative craniofacial morphology. The planned cranial vault and facial bipartition osteotomies and the planned reshaping are also shown. B, Frontal facial views before and after anterior cranial vault and facial bipartition reconstruction. C, Profile views before and after facial bipartition reconstruction. D, Intraoperative views after anterior cranial vault and facial bipartition reconstruction. E, Axial computed tomography scan views through the midorbits before and after reconstruction that demonstrate improvement in orbital hypertelorism and orbital depth with diminished eye proptosis. F, Axial computed tomography scan views through the cranial vault 1 week after facial bipartition (note the dead space in the retrofrontal region) and at 1 year (note that the initial retrofrontal dead space has been resolved by brain expansion). A (right), C (left), E, From Posnick JC: Craniofacial dysostosis. Staging of reconstruction management of the midface deformity, Neurosurg Clin North Am 2:683-702, 1991.
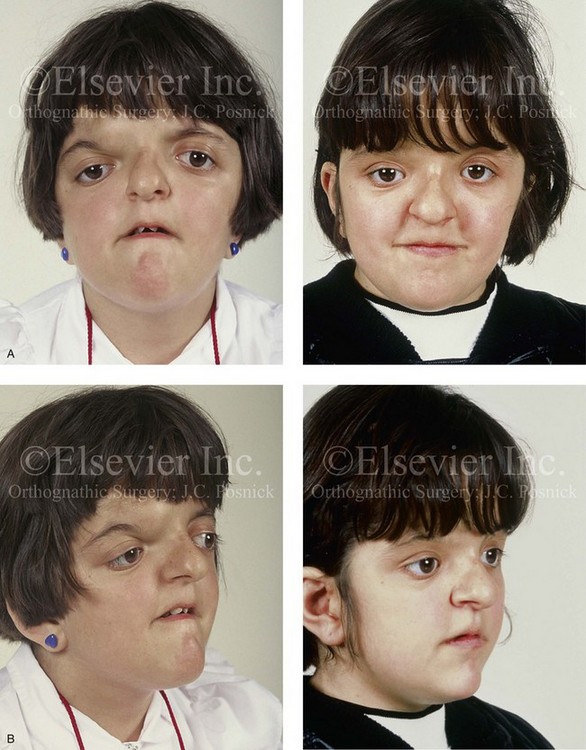
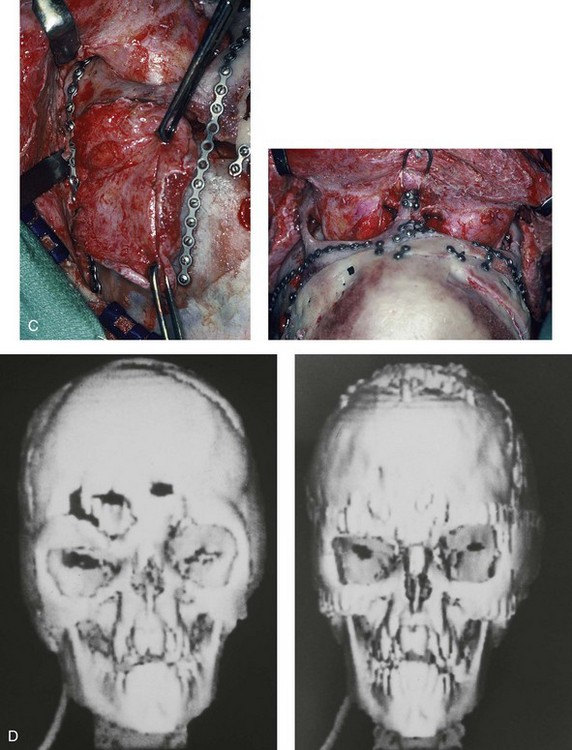
Figure 30-4 An 8-year-old girl who was born with Apert syndrome. She had undergone cranio–orbital surgery on two occasions and an attempted Le Fort III osteotomy at another institution. The previous procedures had resulted in complications, including infection and traumatic encephalocele. She was referred to this surgeon with cranial vault dysplasia, orbital dystopia, hypertelorism, midface deficiency, and skull defects. She then underwent anterior cranial vault, facial bipartition, and additional segmental orbital osteotomies with reshaping and reconstruction. She will require orthodontic treatment and orthognathic surgery during her teenage years to complete the reconstruction. A, Frontal views before and after anterior cranial vault and facial bipartition reconstruction. B, Oblique facial views before and after facial bipartition reconstruction. C, Intraoperative views after anterior cranial vault and facial bipartition reconstruction. Encephalocele and skull defects have also been repaired. D, Computed tomography scan views before and early after reconstruction. From Posnick JC: Craniosynostosis: surgical management in infancy. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1839.
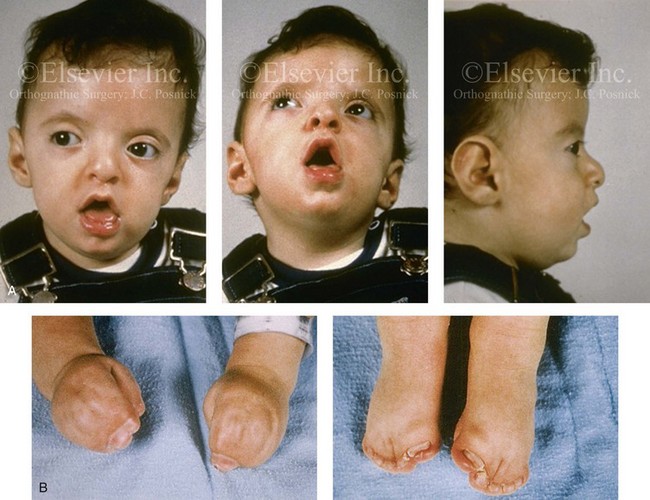
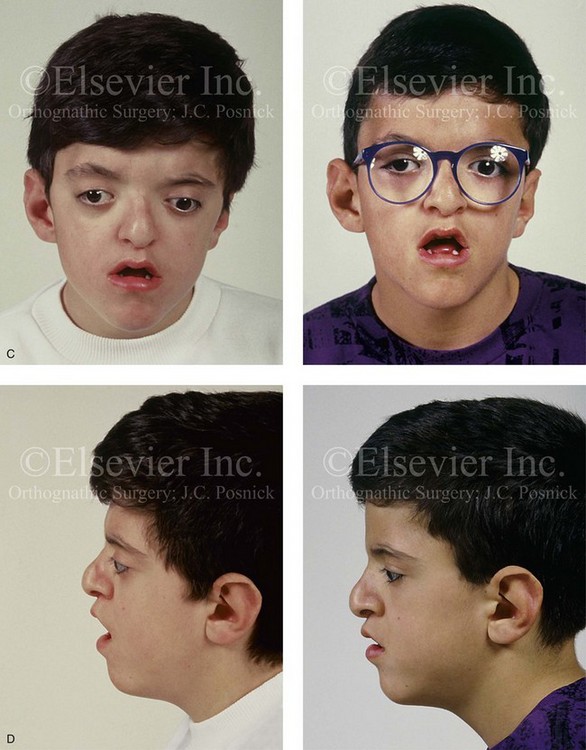
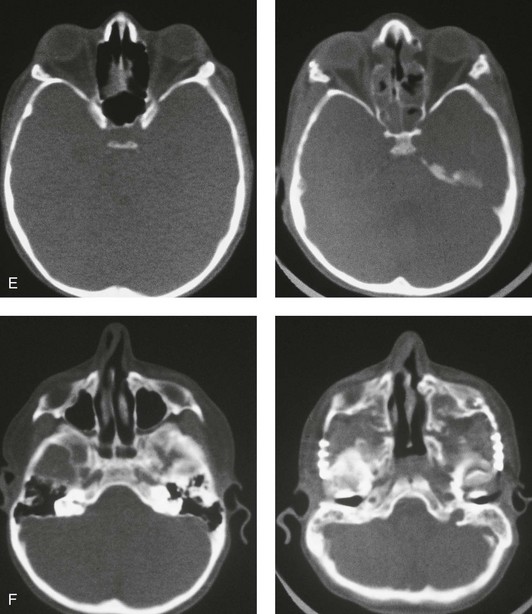
Figure 30-5 A child born with a mild form of Apert syndrome. When he was 10 years old, he was referred to this surgeon and underwent anterior cranial vault and monobloc osteotomies with advancement. He will require orthognathic surgery combined with orthodontic treatment to complete the reconstruction. A, Facial views during infancy. B, Views of the hands and feet that indicate complex compound syndactyly. C, Frontal facial views before and after anterior cranial vault and monobloc osteotomies with advancement. D, Profile views before and after monobloc reconstruction. E, Axial computed tomography scan views through the midorbits before and after reconstruction indicating relief of proptosis. F, Axial computed tomography scan views through the zygomatic arches before and after reconstruction.
Skeletal abnormalities with Apert syndrome include symmetric syndactyly of the hands and feet; abnormal shoulders with significant limitation of motion; and abnormalities of the elbows, hips, knees, rib cage, and spine.46 It should be carefully noted that the radiologic interpretation of the hands, feet, and cervical spine as being affected by so-called “progressive fusion” is very misleading. In actuality, there is a failure of cartilage segmentation during embryonic and fetal life, so these abnormalities are preset (ab initio). These failures in cartilage segmentation cannot be readily observed radiographically at birth. When they do become evident radiographically after birth, they are said to be a “progressive abnormality,” which they are not. The same can be said for the cervical vertebrae,46 as 68% of these patients have cervical abnormalities, particularly involving C5 and C6. Before considering a surgical procedure, magnetic resonance imaging of the brain, radiography of the cervical spine, and the assessment of cardiovascular (10% of patients) and genitourinary (9.6% of patients) anomalies should be carried out.44,46,48
Patients with Apert syndrome also suffer from reduced nasopharyngeal dimensions and reduced patency of the posterior choanae; these features pose the risks of respiratory embarrassment, obstructive sleep apnea (OSA), cor pulmonale, and even sudden death. During infancy, patients may be trialed on a nasopharyngeal airway, but tracheostomy may be the only definitive treatment. Clinicians must always maintain a high index of suspicion for OSA and monitor patients for snoring or an unusual amount of daytime somnolence. When either of these is apparent, patients should be referred to a sleep center for proper diagnosis and treatment. It should be carefully noted that, although patients with Apert syndrome very commonly have OSA, the excessive sweating that occurs with this syndrome is not a sign of OSA but rather a consequence of the increased number of sweat and sebaceous glands that accompany the syndrome. Even those patients who have surgical advancement of the midface in combination with continuous positive airway pressure (CPAP) will still have excessive sweating of the head at night and of the hands all the time. Another consequence of an increased number of sweat and sebaceous glands is a high frequency of conglobate acne during adolescence, often with extension to the forearm, the thighs, and the buttocks. The acne vulgaris of Apert syndrome suggests exquisite end-organ responsiveness to steroid hormones.46
Ocular findings include hypertelorism, proptosis (often asymmetric), and down-slanting palpebral fissures. The presence of V-pattern strabismus, which is marked by divergent upgaze and esotropic downgaze, is common and secondary to structural abnormalities of the extraocular muscles (i.e., the absence of the superior rectus muscle). Hyperopia, myopia, and astigmatism can also be present. Strabismus and significant refractive errors can sometimes cause amblyopia. Proptosis in Apert syndrome may require tarsorrhaphies to prevent exposure keratitis.131
Hearing loss occurs in 90% of these patients, with 80% of cases resulting from conductive pathology. Inner-ear anomalies are found in all patients; these most commonly present as dilated vestibules, malformed semicircular canals, and cochlear dysplasia.285 Otitis media is common, and it is possibly related to the presence of cleft palate and accompanying eustachian tube dysfunction.
Oral and maxillofacial anomalies can also be present. The palate can be highly arched and constricted with a median furrow in up to 94% of patients. The hard palate is often shorter than normal, whereas the soft palate is longer and thicker than normal. Clefting of the soft palate occurs in 41% of patients, and a bifid uvula is present in 35%. Dental anomalies include severely delayed eruption (68%), ectopic eruption (50%), and shovel-shaped incisors (30%). Dental crowding is more severe in the maxilla than in the mandible. Common dental deformities include anterior open bite (73%), posterior crossbite (63%), and mandibular overjet (81%).46,48
Crouzon Syndrome
Crouzon syndrome is characterized by craniosynostosis, maxillary hypoplasia, shallow orbits, and ocular proptosis.* (Figs. 30-6 through 30-11). Most cases are associated with brachycephaly secondary to bicoronal synostosis; however, trigonocephaly, scaphocephaly, and cloverleaf skull have also been noted. Cranial malformation depends on the order and rate of progression of the sutural synostosis. The cranial volume of patients with Crouzon syndrome is much smaller than that found in patients with Apert syndrome. Central nervous system findings include shunted hydrocephalus (25%), chronic tonsillar herniation (72%), jugular foramen stenosis with venous obstruction (30%), seizures (10.5%), frequent headaches (29%), and mental deficiency (3%).46,48 As a result of severe maxillary deficiency, patients should be monitored for snoring and excess daytime somnolence. If this is severe during infancy, tracheostomy may be the only option. Older children should be initially worked up with a polysomnographic study. Treatment options range from CPAP therapy to midface advancement on the basis of the severity level.46
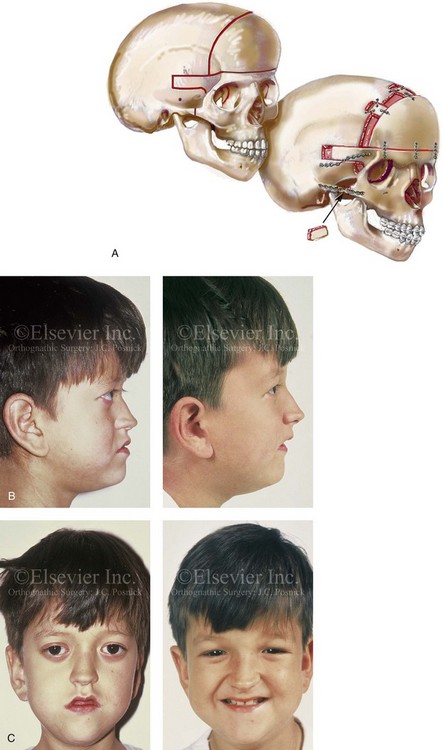
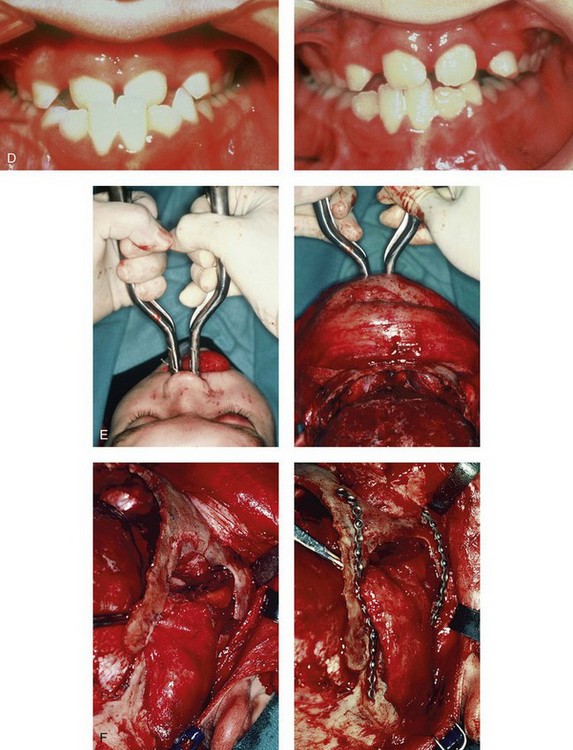
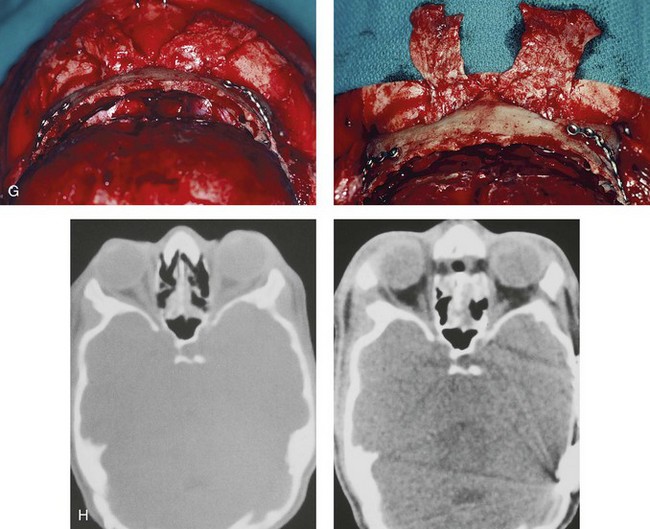
Figure 30-6 An 8-year-old boy who was born with Crouzon syndrome and who underwent a first-stage cranio–orbital procedure when he was 6 weeks old by a neurosurgeon who was working independently. He was referred to this surgeon and then underwent anterior cranial vault and monobloc osteotomies with advancement. A, Illustration of craniofacial morphology before and after anterior cranial vault and monobloc osteotomies with advancement. B, Profile views before and after reconstruction. C, Frontal views before and after anterior cranial vault and monobloc reconstruction. D, Occlusal views before and after reconstruction. Note the improvement but without a full correction of the malocclusion; this will await orthognathic correction during the patient’s teenage years. E, Intraoperative view of disimpaction after monobloc osteotomy with the use of nasomaxillary forceps. F, Intraoperative view of monobloc showing tenon extension and zygomatic arch advancement before and after plate and screw fixation. G, After monobloc advancement and fixation, dead space in the retrofrontal region with communication into the nasal cavity can be seen. Pericranial flaps are elevated from under the surface of the scalp for the closure of cranio–nasal communication. H, Axial computed tomography scan views through the midorbits before and after reconstruction. Reconstruction resulted in increased intraorbital depth and decreased proptosis. A, B, C,D, E, H, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1889.
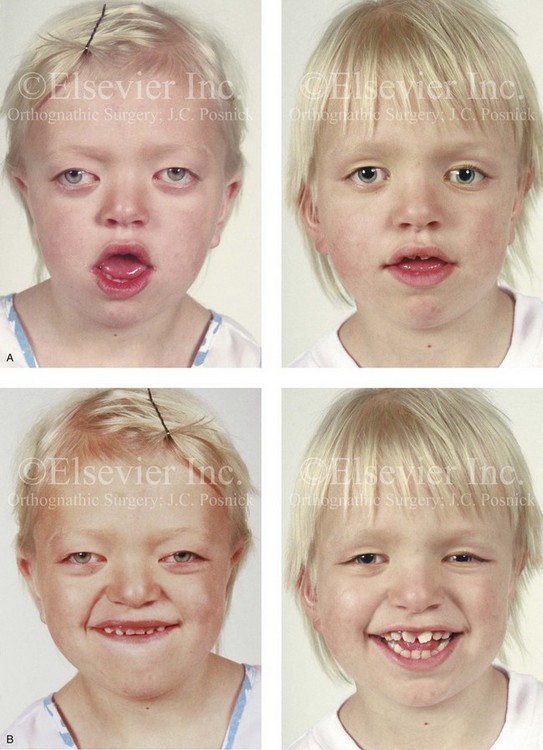
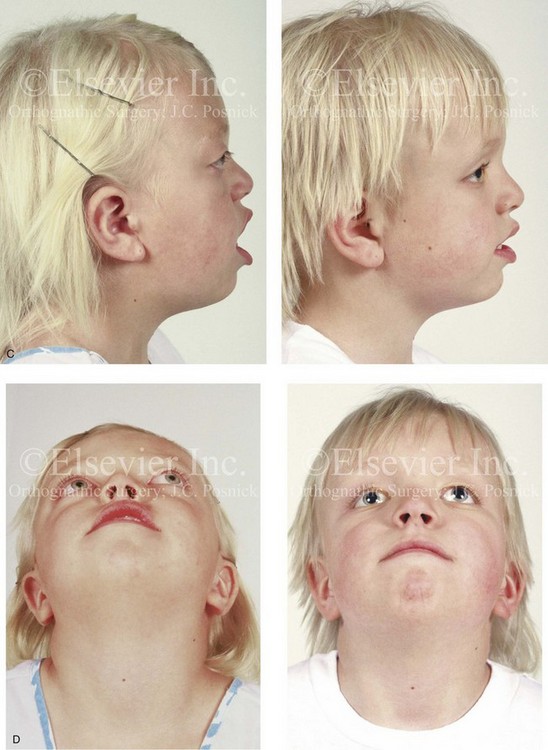
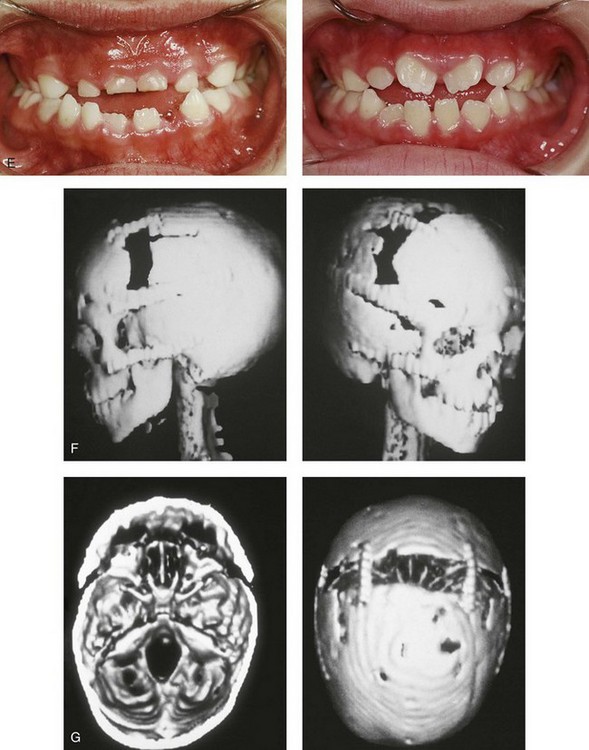
Figure 30-7 A 6-year-old girl who was born with Crouzon syndrome and who underwent anterior cranial vault and monobloc osteotomies with advancement. A, Frontal facial views in repose before and after anterior cranial vault and monobloc reconstruction. B, Frontal views with smile before and after monobloc reconstruction. C, Profile views before and after monobloc reconstruction. D, Worm’s-eye views before and after monobloc reconstruction. E, Occlusal views before and after reconstruction. Note the improvement but without the correction of the malocclusion; this will await orthognathic surgery during the patient’s teenage years. F and G, Computed tomography scan views immediately after reconstruction that indicate advancement with increased volume in the anterior cranial vault and orbits. A, C, D, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.
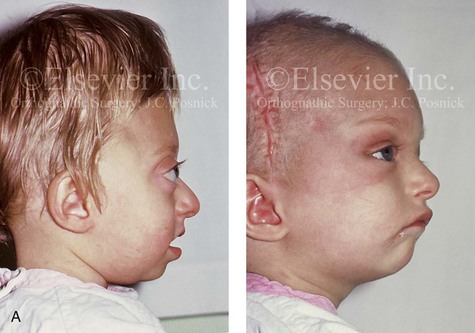
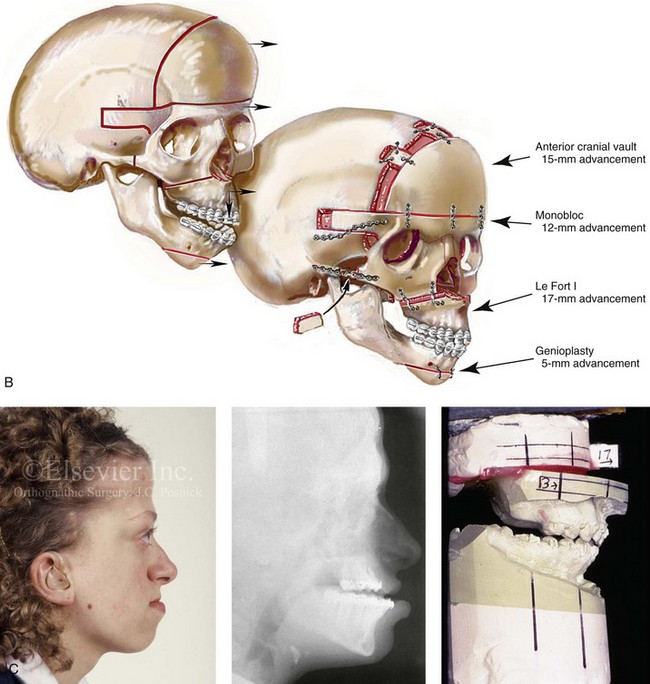
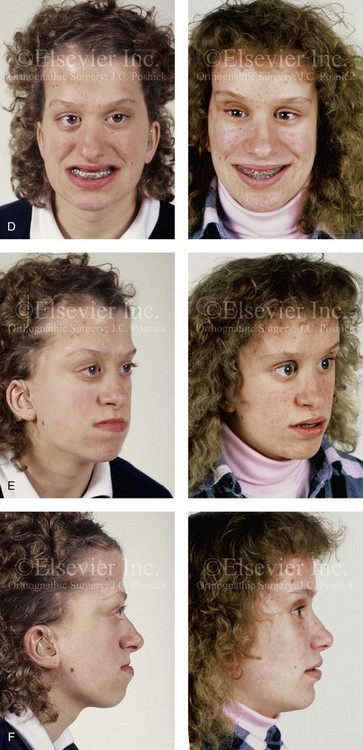
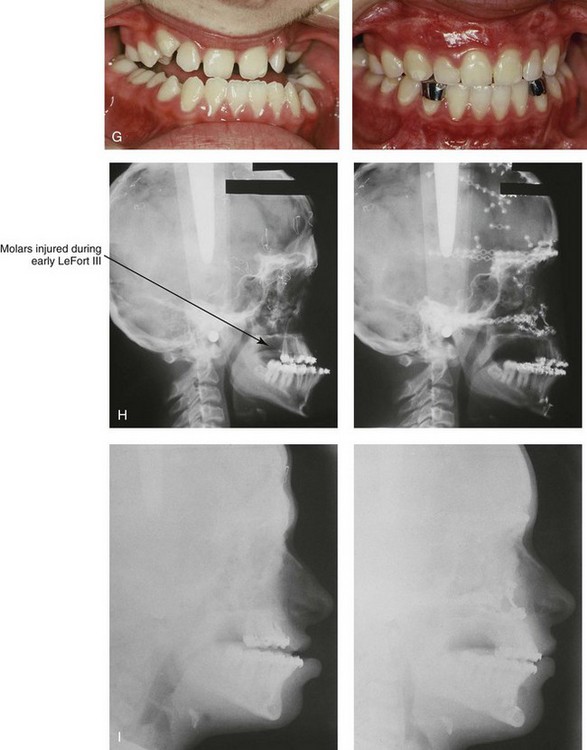
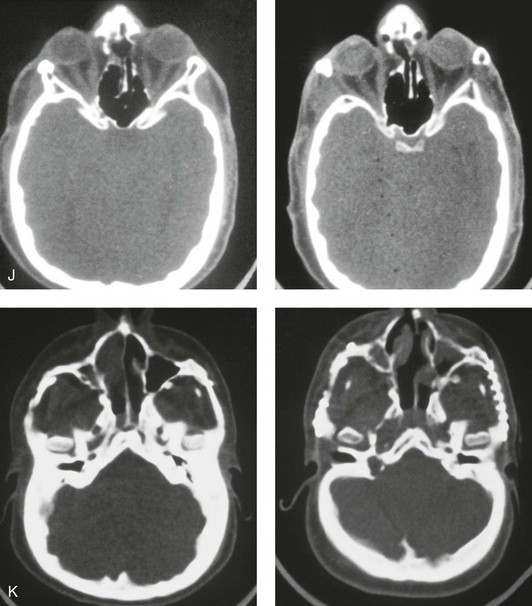
Figure 30-8 A child who was born with Crouzon syndrome and who underwent bilateral coronal suture release when she was 3 months old. Additional craniotomy and cranial vault reshaping were completed when she was 9 months old. When the patient was 2 years old, she underwent a Le Fort III midface osteotomy and forehead advancement procedure through an intracranial approach. She presented to this surgeon when she was 14 years old with residual deformity for which she underwent simultaneous anterior cranial vault, monobloc, Le Fort I, and chin osteotomies with differential advancements. A, Profile facial views at 1 year of age and at 2 years of age after Le Fort III and anterior cranial vault advancement. B, Illustration of planned and completed anterior cranial vault, monobloc, Le Fort I, and chin osteotomies. C, Profile view and lateral cephalometric radiograph before surgery. Articulated dental casts after model planning that indicate that 17-mm advancement is required at the occlusal level. The amount of advancement required at the supraorbital ridge level was just 12 mm. Note the absence of permanent molars in maxilla; this is the result of the Le Fort III osteotomy carried out when the patient was 2 years old. The pterygomaxillary disjunction destroyed the developing teeth. D, Frontal facial views before and after reconstruction. E, Oblique facial views before and after reconstruction. F, Profile views before and after reconstruction. G, Occlusal views before and after reconstruction and orthodontic treatment. H, Lateral cephalometric radiographs before and after reconstruction. I, Soft-tissue lateral cephalometric radiographs before and after reconstruction. J, Axial computed tomography scan views through the midorbits before and after reconstruction confirm relief of proptosis. K, Axial computed tomography scan views through the zygomatic arches before and after reconstruction. A, B (right), D, G, H (left), I, J, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.
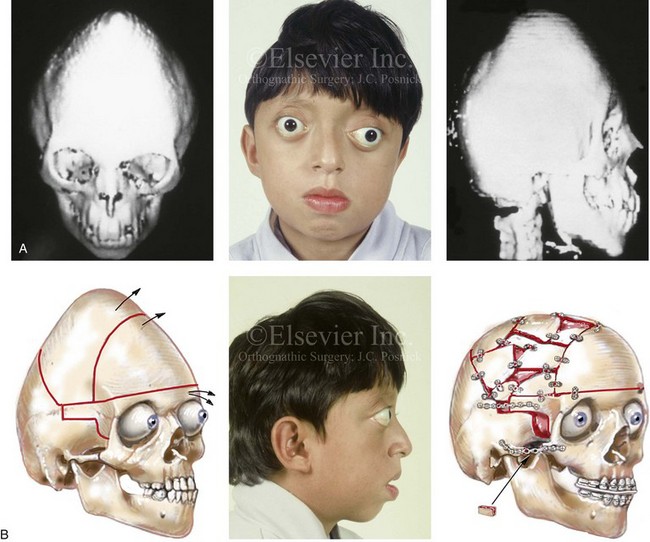
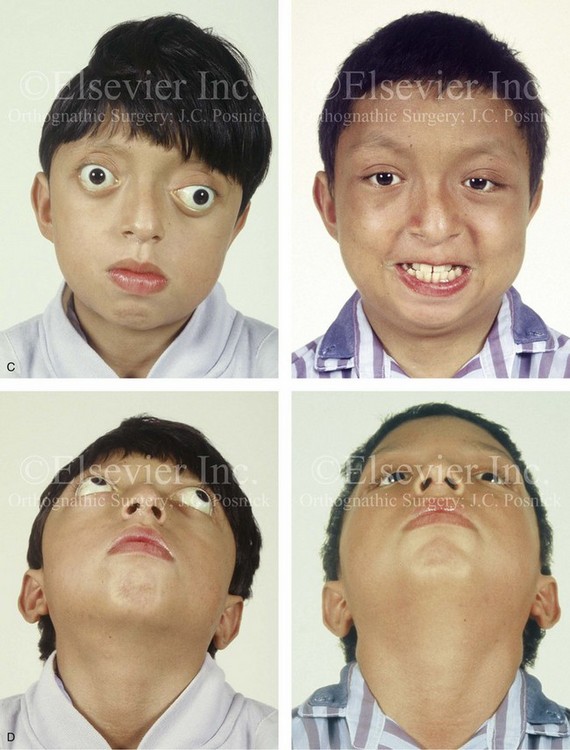
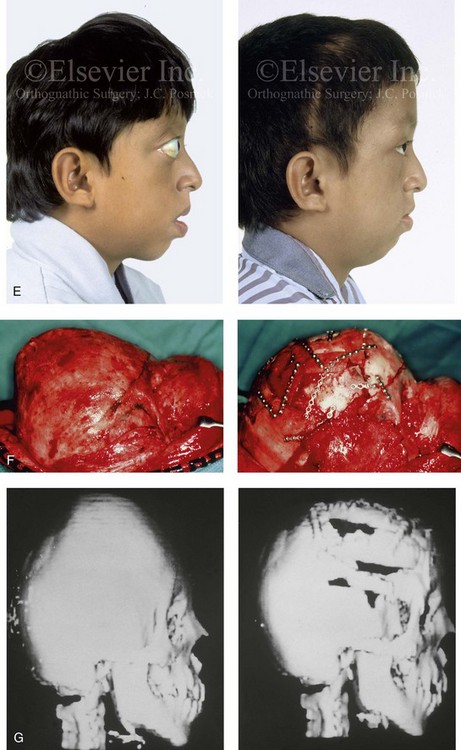
Figure 30-9 A 12-year-old boy with unrepaired Crouzon syndrome was referred to this surgeon for evaluation. He underwent total cranial vault and monobloc osteotomies with reshaping and advancement. A, Facial and computed tomography scan views before surgery. B, Profile facial view before surgery. Illustrations of the patient’s craniofacial morphology with planned osteotomy locations indicated. A second illustration is shown after the proposed osteotomies were completed, with reshaping and advancement. C, Frontal facial views before and after reconstruction. D, Worm’s-eye views before and after reconstruction. E, Profile views before and after reconstruction. F, Intraoperative views before and after total cranial vault and monobloc osteotomy with reshaping and advancement. G, Computed tomography scan views before and immediately after reconstruction. A (right), B (left, right), C (right), D (right), E (right), F, G, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.
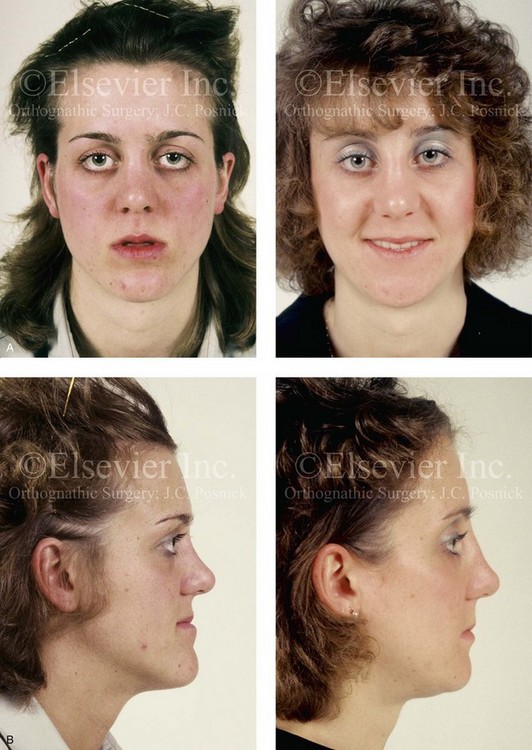
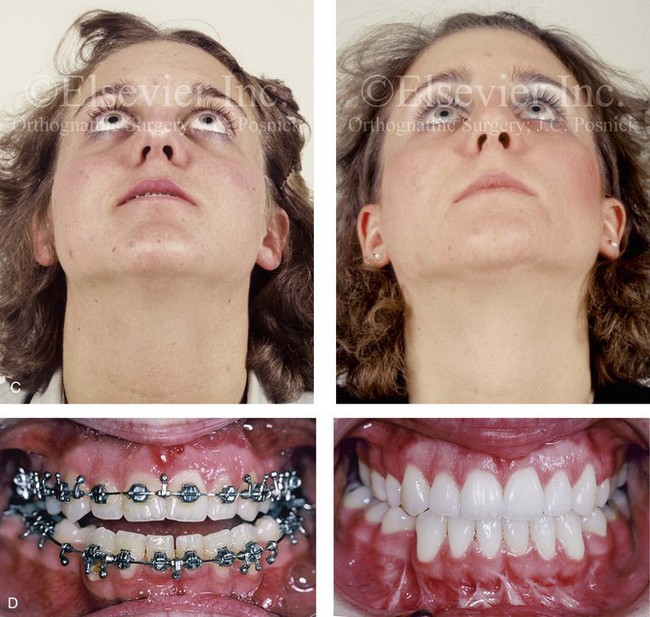
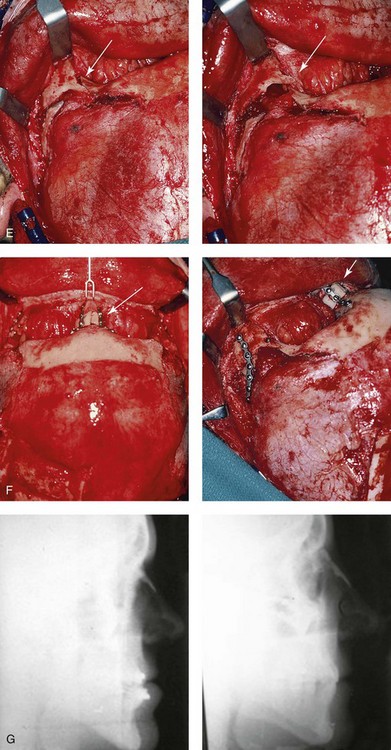
Figure 30-10 A 15-year-old girl with Crouzon syndrome that is characterized by mild to moderate midface deficiency with retrusion of the infraorbital rims, the zygomatic buttresses, and the maxilla. The midface hypoplasia results in increased scleral show, nasal obstruction, and malocclusion. The cranial vault and the upper orbits have normal morphology. Orthodontic camouflage treatment was in progress, including mandibular bicuspid extractions with retraction. The patient was referred to this surgeon for evaluation and treatment. She then underwent a Le Fort III extracranial osteotomy with advancement. A, Frontal facial views before and after reconstruction. B, Profile views before and after reconstruction. C, Worm’s-eye views before and after reconstruction. D, Occlusal views before and after reconstruction. E, Intraoperative view. Note the potential for unsightly step-off at the lateral orbital rim after advancement. F, Cranial bone graft is placed in the frontonasal region. Note the unavoidable lengthening of the nose and the potential for the flattening of the nasofrontal angle that occurs as part of the Le Fort III advancement. G, Lateral cephalometric radiographs before and after reconstruction. A, B (right), C, D, E, F (left), G, from Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.

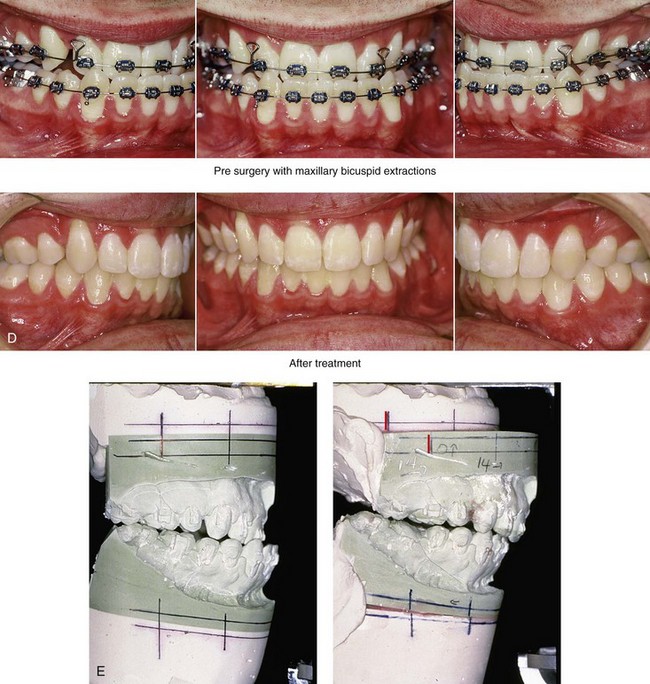
Figure 30-11 A 19-year-old boy who was born with Crouzon syndrome. When he was 11 years old, he underwent a Le Fort III osteotomy with advancement via an extracranial approach by another surgeon. He presented to this surgeon during his late teenage years with asymmetric and dystopic orbits, zygomatic asymmetry, a retrusive upper jaw, an asymmetric lower jaw, and a vertically long chin. He underwent a combined orthodontic (upper bicuspid extractions) and surgical approach. The procedures included Le Fort I osteotomy (horizontal advancement); bilateral sagittal split ramus osteotomies (correction of asymmetry); and osseous genioplasty (vertical reduction and horizontal advancement). He also underwent the reopening of a coronal scalp incision with the harvesting of split cranial grafts and the recontouring and augmentation of the orbits and the zygomas. A, Frontal views before and after reconstruction. B, Oblique facial views before and after reconstruction. C, Profile views before and after reconstruction. D, Occlusal views before and after reconstruction. E, Articulated dental casts that indicate analytic model planning.
Cervical vertebral anomalies are found in 25% of patients with Crouzon syndrome, and all involve C2 and C3. Anomalies should be assessed before the patient undergoes anesthesia for any craniofacial procedure. Patients with Crouzon syndrome already have problematic airways as a result of their relatively inflexible necks; thus, cervical anomalies will compound the issue.46
Ophthalmologic abnormalities include hypertelorism, pronounced ocular proptosis, exotropia, exposure keratitis, poor vision, and optic atrophy. Luxation of the eye globes may be observed in some instances. Although emergency reduction followed by tarsorrhaphies may be necessary, some patients can luxate and reduce all by themselves.131
Oral findings include lateral palatal swellings (50%), obligatory mouth breathing (32%), bifid uvula (9%), and cleft palate (9%). The maxillary dental arch is shortened and associated with a posterior crossbite, dental crowding, and ectopic eruption of the maxillary first molars. Anterior open bite, mandibular overjet, and crowding of the mandibular anterior teeth are also common.46
Pfeiffer Syndrome
Pfeiffer syndrome is characterized by craniosynostosis; midface deficiency; broad thumbs, great toes, or both; brachydactyly; variable soft-tissue syndactyly; and other anomalies (Figs. 30-12 through 30-15).188,231 Three clinical subtypes have been proposed. Type 1 is representative of the aforementioned description. Type 2 is characterized by cloverleaf skull, elbow ankylosis or synostosis, and a cluster of unusual anomalies in addition to the Type 1 characteristics. Type 3 involves a very short cranial base, severe ocular proptosis, elbow ankylosis or synostosis, and an assortment of unusual anomalies. Types 2 and 3 also include an increased risk for neurodevelopmental difficulties. A favorable outcome can be achieved in some cases with aggressive medical and surgical management; however, normal outcome is not the rule, and neurodevelopmental outcome and life expectancy prognoses remain guarded in most cases.44 Although these subtypes are useful, they have limited nosologic status, and some clinical overlap does occur.
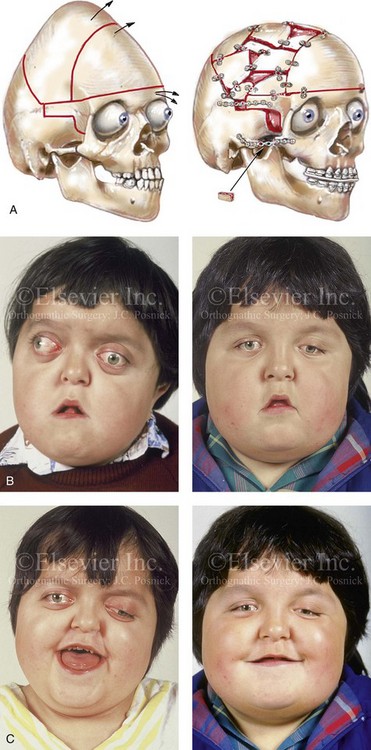
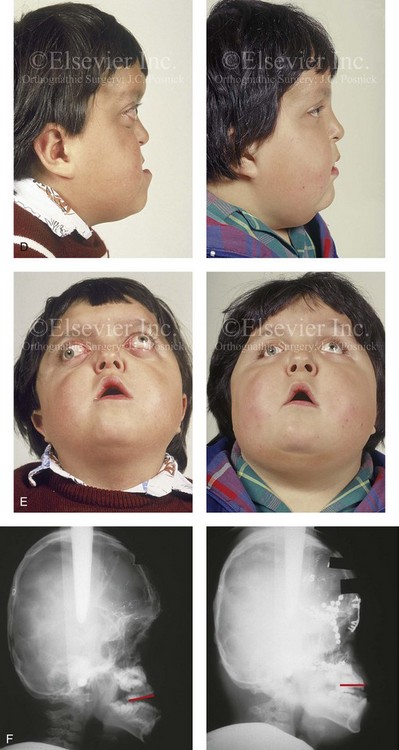
Figure 30-12 A 7-year-old boy who was born with Pfeiffer syndrome was referred for evaluation. He previously had undergone bilateral lateral canthal advancements when he was 3 months old; these were performed by a neurosurgeon who was working independently. The patient then required the placement of a ventriculoperitoneal shunt. He presented to this surgeon with total cranial vault dysplasia, orbital dystopia, and midface deficiency. He underwent cranial vault and monobloc osteotomies with reshaping and advancement. A, An illustration of the proposed cranial vault and monobloc osteotomies and reconstruction is also shown. B, Frontal facial views in repose before and after cranial vault and monobloc reconstruction. C, Frontal facial views with smile before and after reconstruction. D, Profile views before and after reconstruction. E, Worm’s-eye views before and after reconstruction. F, Lateral cephalometric radiographs before and after reconstruction. A (left), B, D, from Posnick JC: Craniofacial dysostosis: staging of reconstruction and management of the midface deformity, Neurosurg Clin North Am 2:683-702, 1991.
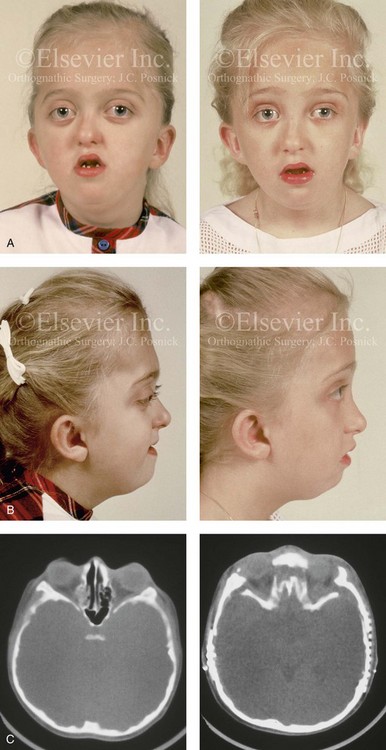
Figure 30-13 A 6-year-old girl who was born with Pfeiffer syndrome. She had undergone cranio–orbital reshaping earlier during her childhood. She presented to this surgeon with a constricted anterior cranial vault, orbital dystopia, and midface deficiency. She underwent anterior cranial vault and monobloc osteotomies with reshaping and advancement. A, Frontal facial views before and after reconstruction. B, Profile views before and after reconstruction. She still requires orthodontic treatment and orthognathic surgery, which are planned for her teenage years. C, Axial computed tomography scan views through the midorbits before and after reconstruction.
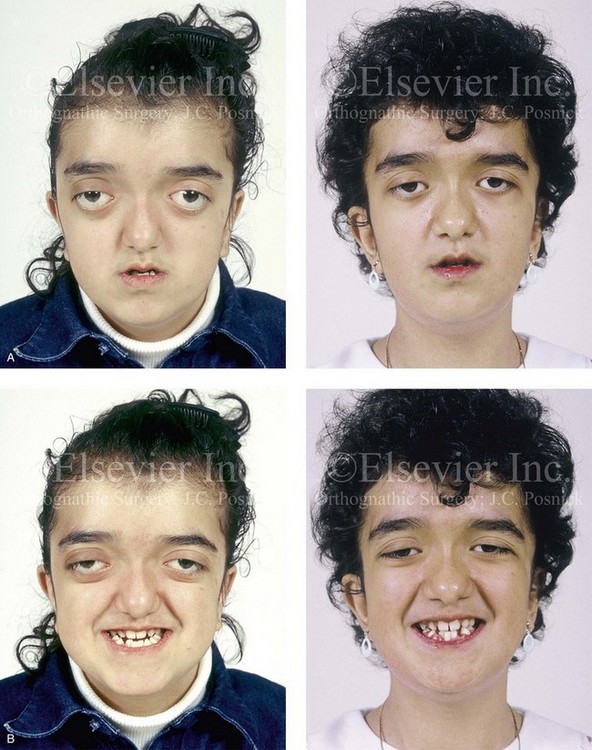
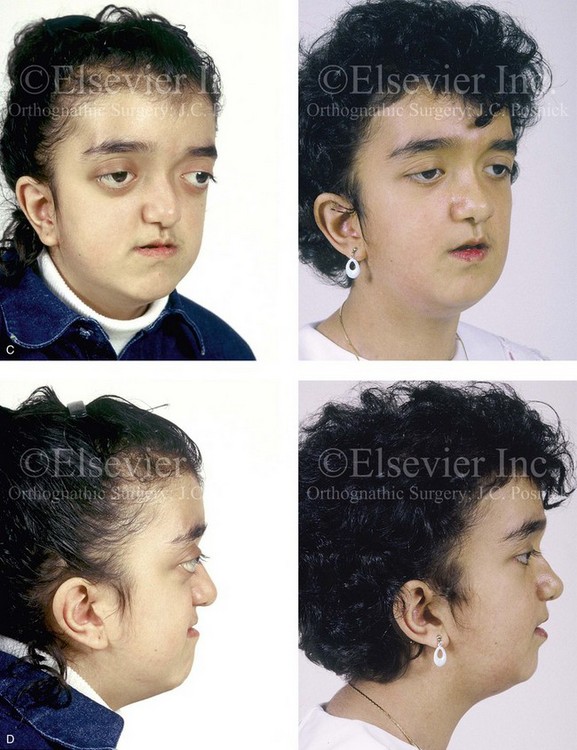
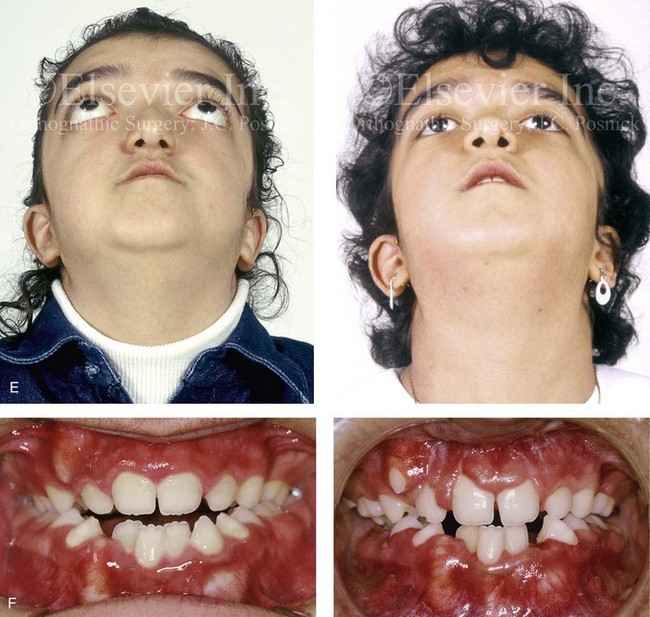
Figure 30-14 A 12-year-old girl who was born with Pfeiffer syndrome was referred to this surgeon for evaluation. She underwent anterior cranial vault and facial bipartition osteotomies with reshaping. She is shown before and after reconstruction. Quantitative analysis of the presenting deformity on the basis of computed tomography scan views confirmed the abnormal cranial vault length (85% of normal), medial orbital wall length (68% of normal), and zygomatic arch length (83% of normal) as well as the extent of globe protrusion (140% of normal). All measurements demonstrated horizontal deficiency in the upper face and the midface. In addition, the anterior interorbital distance (136% of normal), the mid-interorbital distance (129% of normal), the lateral orbital distance (121% of normal), and the intertemporal distance (132% of normal) all indicated a degree of upper-face hypertelorism. The patient then underwent anterior cranial vault and facial bipartition osteotomies for reconstruction. She achieved improved horizontal facial depth, and the upper face hypertelorism was also improved. A, Frontal facial views in repose before and after reconstruction. B, Frontal facial views with smile before and after reconstruction. C, Oblique facial views before and after reconstruction. D, Profile views before and after reconstruction. E, Worm’s-eye views before and after reconstruction. F, Occlusal views before and after reconstruction. Note that the occlusion is improved but that the patient will require orthognathic surgery and orthodontic treatment during her teenage years. From Posnick JC, Waitzman A, Armstrong D, Pron G: Monobloc and facial bipartition osteotomies: quantitative assessment of presenting deformity and surgical results based on computed tomography scans, J Oral Maxillofac Surg 53:358-367, 1995.
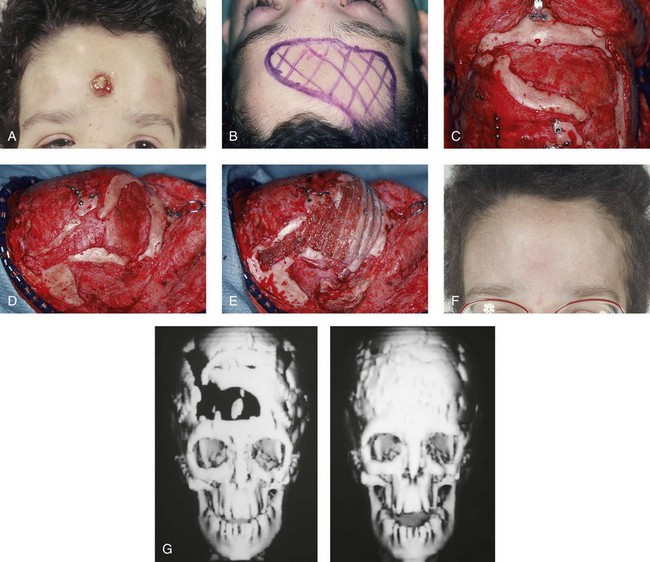
Figure 30-15 A 12-year-old boy with Noonan syndrome. When he was 5 years old, he underwent bilateral total orbital osteotomies with medial translocation for the correction of orbital hypertelorism; this was carried out by another surgeon. When he was 12 years old, he presented to this surgeon with a retruded irregular forehead, shallow orbits, residual proptosis, and midface deficiency. He underwent anterior cranial vault and monobloc osteotomies with horizontal advancement. An occult cerebral spinal fluid leak with drainage through the nose occurred. Eventually, there was erosion through the mid-forehead skin and resorption of a segment of the anterior and lateral cranial vault. One year after the monobloc procedure, the patient was taken back to the operating room for the repair and closure of the cerebral spinal fluid fistula and a cranioplasty that consisted of autogenous split-rib grafts. The cranial vault reconstruction was successful, without further infection or long-term sequelae. A, Frontal view of the upper face 6 months after the initial surgery, with a cerebral spinal fluid fistula through central forehead. B and C, Intraoperative views of the anterior cranial vault documenting the region of bone loss. D, Intraoperative lateral view that shows the area of bone loss. E, Intraoperative lateral view after autogenous split-rib graft reconstruction. F, Frontal view of the upper face 1 year after dura repair and the split-rib reconstruction of the skull defects. G, Computed tomography scan views before and after successful split-rib graft cranioplasty.
Craniofacial features include brachycephaly as a result of bicoronal synostosis, hypertelorism, ocular proptosis, and midface deficiency.44 Distortion ventriculomegaly, progressive hydrocephalus, and cerebellar herniation are common. Progressive hydrocephalus is much more common among patients with Pfeiffer and Crouzon syndromes than it is among those with Apert syndrome. Gyral abnormalities have been observed in some cases.44 Vertebral anomalies occur in 70% of patients, particularly at C2 and C3, but such anomalies may involve other cervical vertebrae as well as the lumbar vertebrae in some cases. 46 As in Apert and Crouzon syndrome, OSA can affect patients with Pfeiffer syndrome. For a further understanding of diagnosis and therapy, please refer to the relevant discussions in the sections of this chapter about the Apert and Crouzon syndromes. Other anomalies have been noted in some cases, including cardiovascular defects, gastrointestinal issues, and genitourinary anomalies.44
Saethre–Chotzen Syndrome
Saethre–Chotzen syndrome is characterized by a heterogeneous phenotypic presentation that involves craniosynostosis, a low-set frontal hairline, facial asymmetry, ptosis of the eyelids, a deviated nasal septum, brachydactyly, partial soft-tissue syndactyly of the second and third fingers, and various skeletal anomalies.4,33,40,234 Craniosynostosis is facultative and not obligatory (i.e., some patients do not have it). When it is present, the time of onset and the degree of craniosynostosis can be quite variable. Brachycephaly is found most commonly, and this is followed by synostotic anterior plagiocephaly. Frontal bossing, parietal bossing, and occipital flattening can also accompany the cranial deformity. Large late-closing fontanelles and large parietal foramina have also been reported. Ptosis of the eyelids, hypertelorism, and strabismus are common. The ears may be low set, small, and posteriorly angulated, and mild conductive hearing loss is common. Oral anomalies include a narrow or highly arched palate, a cleft palate on occasion, malocclusion, and supernumerary teeth.40
Carpenter Syndrome
Carpenter syndrome is characterized by craniosynostosis (commonly, but not always); preaxial polydactyly of the feet; brachydactyly, clinodactyly, or both; congenital heart defects (these are seen in 33% of patients), such as ventricular septal defect, atrial septal defect, patent ductus arteriosus, pulmonary stenosis, and tetralogy of Fallot; short stature, with a height that is usually below the 25th centile; obesity; and mental deficiency in some cases.28,39 Craniosynostosis involves the sagittal and lambdoid sutures first, with the coronal suture being the last to close. The head is usually broad, but it is variable in shape, and asymmetry may be a feature. Other features include dystopia canthorum, down-slanting palpebral fissures, epicanthal folds, low-set ears, and a short neck. The mandible may be hypoplastic, and the palate may be narrow or highly arched.39
Neurologic Aspects of Craniosynostosis
Generally, craniosynostosis tends to restrict intracranial volume. The earlier synostosis occurs, the more dramatic the effect on subsequent cranial growth and development. The later synostosis occurs, the less the effect on cranial growth and development. Growth restriction also worsens with increasing sutural involvement; complex craniosynostosis with two or more affected sutures demonstrates higher growth restriction as compared with single sutural involvement. There are two exceptions to this observation: scaphocephaly and Apert syndrome. With scaphocephaly, mechanical forces produce a large head circumference; with Apert syndrome, brain weights have been shown to be above the 97th centile, regardless of age.46,48
Mental deficiency is correlated with the number of fused sutures, and it occurs more frequently with two or more sutural involvements than with single-suture synostosis. In addition, mental deficiency occurs more commonly with coronal synostosis than with other single-suture craniosynostoses. With single-suture craniosynostoses (other than coronal), mental deficiency may be more attributed to a brain malformation rather than to growth restriction. This may occur in patients with trigonocephaly and even in some cases of sagittal synostosis.25
In the section about Apert syndrome, a distinction was made between benign megalencephaly or distortion megalencephaly and progressive hydrocephalus. The mechanism of hydrocephalus may vary. Although communicating hydrocephalus with obstruction at the level of the basal cisterns is most common, aqueductal stenosis is fairly frequent. Chronic tonsillar herniation occurs in 73% of patients with Crouzon syndrome as compared with only 1.9% of patients with Apert syndrome.62 Premature synostosis of the lambdoid suture may be responsible for this finding. It has also been suggested that the obstruction of the venous drainage of the brain may play a role by narrowing the jugular foramina. Some patients are able to compensate well for their hydrocephalus.25
Clinicians must maintain a high index of suspicion for hydrocephalus in all patients with craniosynostosis, particularly those with complex or syndromic types.77,91,108,170,174 Among the syndromic craniosynostoses, shunted hydrocephalus is found in 26.6% of patients with Crouzon syndrome and in 27.8% of patients with Pfeiffer syndrome but in only 6.5% to 8% of patients with Apert syndrome.46 Increased intracranial pressure in craniosynostosis may be the result of a mismatch in cranial volume to brain growth or of hydrocephalus. Increased intracranial pressure is found most commonly in patients with multiple suture synostoses.9,26,49,58,122,191,222,226,227–230,245,266 However, recent studies have indicated that increased pressure may also occur in patients with single-suture synostosis. In one report, 3 out of 18 children with scaphocephaly had increased intracranial pressure during 12- to 24-hour recordings.265
When pressure is low grade, clinical symptoms can be subtle or absent. Headache is the classic symptom that is associated with increased intracranial pressure of any cause. However, children seem to experience this inconsistently, and infants are not developed enough to communicate their symptoms. The most common physical finding is papilledema, although this may not be as apparent during infancy or early childhood, when bulging may occur at the anterior fontanel instead. Longstanding papilledema may result in optic atrophy and eventual blindness.25
Regardless of the cause, increased intracranial pressure tends to rise during rapid eye movement sleep, presumably as a result of increased cerebral blood flow. Indeed, plateaus of increased intracranial pressure that last approximately 10 to 120 minutes have been observed during sleep. Conversely, intracranial pressure may be normal while the patient is awake or between periods of rapid eye movement sleep. Thus, the diagnosis of increased intracranial pressure may be missed by a single lumbar puncture measurement. Reasonably safe ways of long-term, direct measurement of increased intracranial pressure are possible with the use of an epidural pressure transducer to record pressure for at least 12 hours. In this way, the significance of increased intracranial pressure as a contributing factor to mental deficiency can be examined.25
With these considerations in mind, the neurologic assessment of patients with craniosynostosis can be performed as follows. The medical history should concentrate on genetic factors as well as the on child’s development. The physical examination should include a careful search for associated anomalies, with scrupulous attention paid to each of the cranial nerves (and particularly to cranial nerves II and VIII). Head circumference should be plotted on the standard growth curve. Investigations should include a skull radiograph, head computed tomography (CT) scan, brain magnetic resonance imaging, initial assessment for hydrocephalus, and formal assessment for hearing. An ophthalmologic examination is mandatory. Depending on the findings, a 12- to 24-hour pressure recording should be considered. Whenever possible, a formal developmental or newborn psychologic battery should be administered before surgery. The family should be assessed for psychosocial problems, especially when a complicated course of surgical interventions is contemplated. Follow up by a pediatrician or pediatric neurologist at regular intervals is essential. Assessments at monthly intervals for the first 12 months, every 3 months during the second year, and every 6 months until 6 years of age seem appropriate.25
Effects of Midface Deficiency on the Upper Airway
Sleep apnea may be central, obstructive, or of mixed origin; proper workup and assessment are crucial to the establishment of the correct diagnosis and treatment.* Central apnea may result from intracranial hypertension. If so, the condition should improve upon brain decompression by appropriate cranio–orbital or posterior cranial vault expansion or when effective shunting of the hydrocephalus is accomplished.
OSA has already been addressed in the sections about the Apert, Crouzon, and Pfeiffer syndromes.105,138,149,190 OSA in a child who is affected by one of these conditions is frequently treated with tracheostomy. It may also be treated by adenoidectomy, tonsillectomy, midface advancement, or CPAP. If left untreated or ineffectively treated, OSA will result in disabilities that include failure to thrive, recurrent upper respiratory infection, cognitive dysfunction, developmental delay, cor pulmonale, and sudden death.173 Midface hypoplasia in the setting of a craniosynostosis syndrome is often a primary cause of OSA. If feasible, surgical midface advancement is the preferred biologic treatment approach; even with such treatment, some patients will still require a tracheostomy or CPAP therapy.
Al-Saleh and colleagues completed a retrospective review of children with syndromal craniosynostosis who were referred to the Hospital for Sick Children in Toronto, Canada, between 1996 and 2008 for initial polysomnography to rule out sleep-related disordered breathing.2 The research confirmed a high prevalence of this type of breathing in children with Crouzon, Apert, Pfeiffer, and Saethre–Chotzen syndromes (N = 35). Despite a spectrum of surgical and non-surgical interventions having been carried out, the complete resolution of the sleep-related disordered breathing could not be routinely achieved.
Ishii and colleagues used lateral cephalometric radiographs to evaluate the nasopharyngeal airway after Le Fort III advancement in patients with Apert or Crouzon syndrome and OSA.114 The researchers documented improvement in the nasopharyngeal airway space after successful midface advancement surgery. Arnaud and colleagues reviewed respiratory improvement in patients with syndromic craniosynostosis and OSA after monobloc (MB) advancement.8 Eighty-eight percent of patients (n = 16) showed improvement as measured by oxygen level during sleep; 4 of 6 patients underwent successful tracheostomy decannulation. In one patient, a tracheostomy was required 6 months after midface advancement for severe and recurrent OSA.
Twenty-four months after MB advancement, Witherow and colleagues documented the significant improvement of airway obstruction on polysomnography in all patients who had been diagnosed with Apert, Crouzon, or Pfeiffer syndrome.280 Before MB advancement, 14 patients were managed with tracheostomy or CPAP therapy for severe OSA. Of these 14 patients who were undergoing MB advancement for severe OSA, only 43% (n = 6) had resolution of their OSA; 8 of 14 patients (57%) remained dependent on tracheostomy or CPAP despite MB advancement.
Nelson and colleagues studied 18 patients with craniosynostosis syndromes who also had coronal synostosis and midface deficiency.172 Eighty-three percent (16 of 18 patients) were treated with tracheostomy or CPAP for known OSA before midface advancement. After the midface advancement procedures, five of the tracheostomy patients were decannulated; in six others, CPAP was no longer required. This represents a 73% (11 of 15 patients) initial success rate for the relief of OSA. The mean postsurgical follow up was 3 years.
Morphologic Considerations
Examination of the patient’s entire craniofacial region should be meticulous and systematic. The skeleton and the soft tissues are assessed in a standard way to identify all normal and abnormal anatomy.65,66,68,202,205,206,211,212,216,217,272,273 Specific findings tend to occur in particular malformations, but each patient is unique. The achievement of symmetry, normal proportions, and the reconstruction of specific aesthetic units is essential to forming an unobtrusive face in a child who is born with one of the craniosynostosis syndromes.
Surgical Approach to Craniosynostosis Syndromes
Historical Perspectives
As early as 1890, Lannelogue134 and then Lane133 described a surgical approach to the treatment of craniosynostosis. Lannelogue’s aim was to remove the fused suture (i.e., strip craniectomy) in the hope of controlling the problem of brain compression within a congenitally small cranial vault. By the turn of the century, Harvey Cushing, who was the most prominent neurosurgeon of his day, suggested that surgical intervention for the problem of craniosynostosis was misdirected and that more attention should instead be given to the schooling of these children.55 Shillito disagreed with Cushing and enthusiastically supported the concept of surgical intervention to improve the outlook for these children.243 He believed that the linear “strip” craniectomy of the fused sutures would “release” the skull and allow the cranium to reshape itself and continue to grow in a normal and symmetric fashion. The strip craniectomy procedures were supposed to allow for a new suture line at the site of the previous synostosis. With the realization that this goal was not biologically feasible, attempts were made to remove portions of the cranial vault surgically and leave large open areas or to use the removed segments as free grafts to refashion the cranial vault shape. Problems with these methods included uncontrolled postoperative skull molding, reossification into dysmorphic configurations, and the occurrence of large residual skull defects.
The concept of the simultaneous release of the fused suture in combination with more meticulous cranial vault reshaping in infants was initially suggested by Rougerie and colleagues232 and then refined by Hoffman and Mohr in 1976 for children who were born with unilateral coronal synostosis.107 In 1977, Whitaker and colleagues proposed a more formal anterior cranial vault and orbital reshaping procedure for unilateral coronal synostosis.279 Marchac and Renier published their experience with a “floating forehead” technique to manage craniosynostosis during infancy; this consisted of simultaneous unilateral coronal and bilateral coronal suture release and forehead and upper orbital osteotomies with advancement.144 Unfortunately, the “floating forehead” technique resulted in unpredictable reossification of the open cranial vault areas.145 In addition, bitemporal constrictions and bulging of the skull concavities were frequent occurrences as reossification occurred. In any case, the hope for midface growth did not materialize.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


