Chapter 3
The Role of the Host Response
Aim
No text in periodontology can avoid discussing the inflammatory and immune responses: subjects which confuse and thus bore many practitioners, often appearing to have little practical utility in the dental surgery. This chapter is the most complex in the book, but attempts to provide a substantially visual guide to key processes that govern disease development and hence therapeutic strategies. The chapter is essential to understanding the remainder of the text, and whilst contemporary, it must also prepare readers for new diagnostic and treatment strategies, some of which are already in the high street.
Outcome
The outcome of reading this chapter will be that the previously mystical world of periodontal immunopathology will make some sense. The reader will also be able to make informed decisions about whether he/she feels certain newer diagnostic tests and therapeutic strategies, whilst biologically effective, are worthwhile, in financial and logistical terms, for their individual patient.
The host-response to microbial plaque is designed to be protective, but the balance is a delicate one where under-activity or indeed over-activity of specific aspects of the response can lead to tissue destruction (Fig 3-1). For example, the patient in Fig 3-2 suffers from the defective function of an enzyme called C1-esterase inhibitor. The enzyme inhibits the activation of complement component C1 (which propagates inflammatory processes). When plaque accumulates at the gingival margin in her mouth, the resulting inflammation and swelling are exaggerated and cause severe gingival oedema and bone loss. There are two main components of the host-response to plaque:
-
natural or innate immunity
-
acquired or specific immunity.
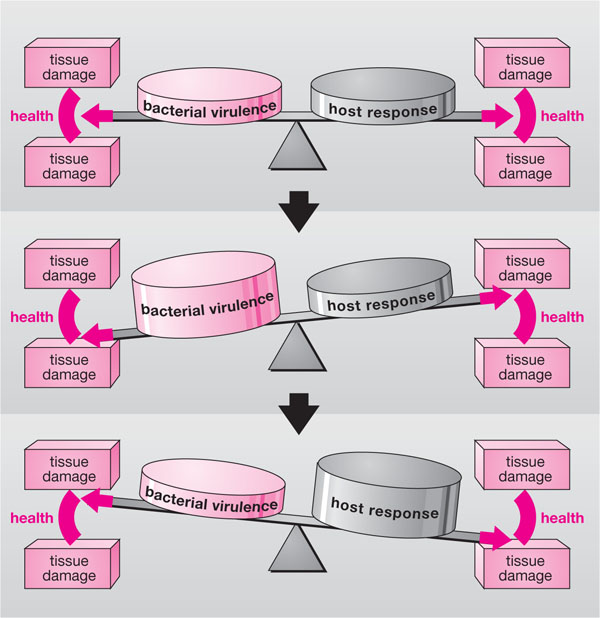
Fig 3-1 The delicate balance between parasitic activity and host defences, which maintain periodontal tissue health. Such equilibrium may easily be disrupted, leading to inflammation and tissue damage.
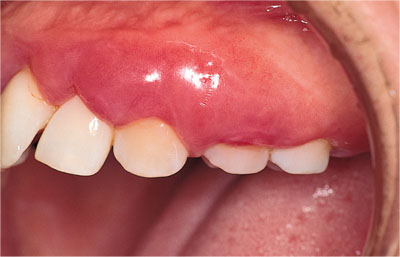
Fig 3-2 Clinical photograph of exaggerated inflammation in a patient with excellent oral hygiene, who suffers from C1-esterase inhibitor dysfunction.
Both types of immunity are involved in the host’s response to plaque and occur at the same time, but the innate system is constantly in function and more rapidly mobilisable, whereas the acquired system involves lymphocytes and specific cell–cell interactions which take more time. Early gingival inflammation largely involves the innate system, the acquired system becoming involved in moderate-to-advanced gingivitis and in periodontitis (i.e. longer-term, chronic disease).
Innate Immunity
This system is the first line of defence. Throughout evolution, it has been highly conserved, and, whilst rapid, it remains crudely indiscriminate relative to the specificity of the acquired system. In terms of periodontal immunology, it is akin to hitting a small nail with a sledgehammer, and hence, when something goes wrong with this system, the collateral tissue damage is significant. Innate immunity involves:
-
intact epithelial barriers
-
lubrication of epithelium with fluids (saliva, GCF) containing anti-bacterial factors
-
the complement cascade
-
cell-signalling molecules called cytokines (e.g. the interleukin family) and chemokines (molecules produced by one cell, which recruit specific leucocytes)
-
vasoactive peptides (like histamine) released from mast cells
-
adhesion molecules
-
PMNL (neutrophil) defence systems
-
macrophages – which also function as antigen presenting cells (APCs) at the beginning of specific immune responses.
Epithelial Barriers
Epithelial barriers were discussed in Chapter 1. The unique nature of the JE, its rapid rate of turnover and permeability to GCF and PMNLs passing through, provide primary first-line defence strategies. The epithelial cells also release cell-signalling molecules (see below) to help start the inflammatory process.
Fluid Lubrication
Whole, mixed saliva contains anti-bacterial enzymes, like lysozyme, and immunoglobulins (antibody) like IgA and IgG, which bind to bacteria. GCF carries all components of serum, including complement and immunoglobulin.
Complement Cascade
The complement cascade is a series of 20 serum glycoproteins, which circulate in inactive forms in the blood stream. When activated, complement components have profound and powerful effects in stimulating inflammation (pro-inflammatory effects). The main role of complement activation is:
-
recruitment of more phagocytes to the area of infection
-
to facilitate binding of phagocytes (e.g. PMNLs) to bacteria, thereby aiding phagocytosis – a process called opsonisation
-
to cause bacterial killing (cell lysis).
There are two pathways for complement activation (Fig 3-3):
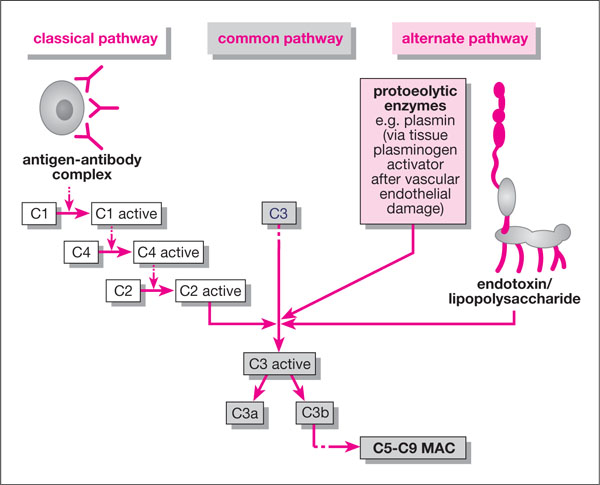
Fig 3-3 The complement cascade.
-
alternative pathway – activated directly by bacterial endotoxin (LPS) when it enters the periodontal tissues and from the cementum reservoir. Other proteolytic enzymes like plasmin and some neuropeptides (e.g. β -endor-phin, released during stress responses) can also activate the alternative pathway.
-
classical pathway – only activated by the formation of antigen-antibody complexes (i.e. after initiation of the acquired immune response).
Both pathways stimulate the activation of complement component C3, which leads to the activation of C5 and the amplification cascade which forms C5-C9, the so-called membrane attack complex (MAC). The methods by which active forms of C3 to C9 recruit phagocytes and propagate inflammation are summarised in Table 3-1. It can be seen how endotoxin can directly activate this cascade and, via complement component C3b, mediate the crude adherence of PMNLs to bacteria (opsonisation). PMNL-killing of the bacteria then follows through extracellular or intracellular killing mechanisms (see later), which may not involve lymphocytes and specific immunity. This type of action is believed to be responsible for early gingivitis.
| Complement Component | Inflammatory Effect |
| C3a and C5a | ↑ blood vessel permeability via mast cell degranulation and release of histamine |
| C3b and C5a | Production of oxygen radicals by leucocytes |
| C5a | Leucocytes stick (marginate) to blood vessel walls Neutrophils release enzymes and oxygen radicals (called degranulation) |
| C3a | Chemotaxis of phagocytes (chemical attraction) |
| C3b | Cytokine (from lymphocytes) production Macrophage secretion Opsonisation (attraction and adhesion of phagocytes) |
| C5a and C567 complex | Leucocyte chemotaxis (movement towards infection) |
| C5b, C6, C7, C8 and C9 | Lysis (splitting open) of gram-negative bacteria Host cell destruction |
Cell-signalling Molecules
Several families of these potent molecules exist; some stimulate cells to release other molecules (cytokines), some attract cells to areas of infection (chemokines) and others stimulate cells to perform other functions (cytokines and lymphokines). The function and dynamics of these highly complex networks are poorly understood and many have seemingly conflicting activities, whilst others have common activities. The main cytokines and their functions in periodontal inflammation are listed in Table 3-2. One of the most important cytokines in periodontal disease is interleukin-1 (IL-1), as it causes bone resorption and is powerfully pro-inflammatory. The genes that code for IL-1 have different forms (polymorphisms) and Kornman and colleagues found that patients who suffered from advanced chronic periodontal disease possessed a polymorphism of IL-1 that resulted in overproduction. A “periodontitis susceptibility test” (PST) has been developed from this work, which identifies from a finger-prick blood sample the presence of the “risk” polymorphism. However, patients’ smoking habits confound the results, and there is no clear evidence that a PST-positive result will mean the patient concerned will develop periodontal disease. In addition, a study in the UK failed to demonstrate the same results as the United States study in a UK cohort.
| Cytokine | Cell of Origin | Effects Upon Inflammatory/ Immune Response |
| Interleukin 1
(IL-1) |
macrophage (MØ), fibroblast, monocytes upon stimulation by endo/exotoxins epithelial cells |
activation of osteoclasts
↑ PMNL margination ↑ prostaglandin PGE2 by fibroblasts |
| Interleukin 6 |
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


