Chapter 3 The resident oral microflora
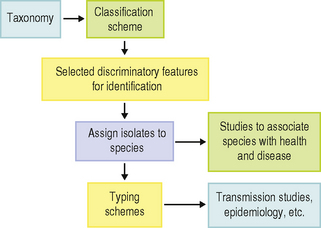
Before the microbial community at individual sites in the mouth can be considered in detail (Chs 4 and 5), the types and properties of the organisms found commonly in health and disease will be described. First, however, it may be instructive to discuss the principles of microbial classification and identification, and describe briefly some of the methods used. Classification is the arrangement of organisms into groups (taxa) on the basis of their similarities and differences. In contrast, identification is the process of determining that a new isolate belongs to a particular taxon; the aim of classification is to define these taxa at the genus or species level. Traditionally, a hierarchical system has existed for the naming of bacteria so that groups of closely-related organisms form a species, and related species are placed in a genus, etc. (Table 3.1); species are designated by Latin or latinized binomials (e.g. Streptococcus mutans; the genus is ‘Streptococcus’ and the species is ‘mutans’, Table 3.1). If an isolate does not belong to an existing taxon, then a new species can be proposed. The naming of bacteria to reflect this classification (nomenclature) is regulated by international committees. Once an organism has been placed in a species, it may be possible to sub-type individual strains; this can be valuable in epidemiological studies investigating transmission of strains between individuals. The interrelationships between these approaches (classification, identification, strain typing) are shown in Fig. 3.1. The classification, nomenclature and identification of microorganisms is referred to as taxonomy, although, sometimes, the terms classification and taxonomy are used interchangeably.
| Taxonomic rank | Example |
|---|---|
| Kingdom | Procaryotae |
| Division | Firmicutes |
| Sub-division | low G+C content of DNA |
| Order | – |
| Family | Streptococcaceae |
| Genus | Streptococcus |
| Species | Streptococcus mutans |
| Serotype* | Streptococcus mutans serotype c |
| Strain* | Streptococcus mutans NCTC 10449** |
* These ranks are not formally recognised in taxonomy, but are of great practical importance.
** NCTC = National Collection of Type Cultures.
Principles of microbial classification
As stated above, the purpose of classification schemes is to develop a logical arrangement of organisms based on their similarities and relationships. This requires the determination and comparison of as many characteristics as possible, although in identification schemes, only a few key discriminatory tests may be needed to distinguish between certain organisms. Early classification schemes relied heavily on morphological and simple physiological criteria such as the shape of the cell, and the pattern of fermentation of simple sugars (Table 3.2). In effect, these approaches analysed only a fraction of the components encoded by the genetic material of the cell (the genome). Chemotaxonomy, in which there is a broader analysis of more complex components of the cell (for example, the chemical composition of the cell wall or of membrane lipids, whole cell protein profiles, etc) led to major improvements in classification schemes. The structure of cells can also be compared using immunological techniques (serology), in which specific antibodies (polyclonal or monoclonal) are used to detect cell surface antigens.
Table 3.2 Some characteristics used in microbial classification and identification schemes
| Characteristic | Examples |
|---|---|
| Cellular morphology | Shape; Gram stain reaction; flagella; spores; size |
| Colonial appearance | Pigment; haemolysis; shape; size |
| Carbohydrate fermentation | Acid or gas production |
| Amino acid hydrolysis | Ammonia production |
| Pattern of fermentation products | Butyrate; lactate; acetate |
| Preformed enzymes | Glycosidases (e.g. α-glucosidase) |
| Antigen | Monoclonal/polyclonal antibodies to cell surface proteins |
| Lipids | Menaquinones, long-chain fatty acids |
| DNA | Base composition (G + C ratio); 16S rRNA gene sequence |
| Enzyme profile | Presence/absence; electrophoretic mobility |
| Peptidoglycan | Amino acid composition e.g. lysine |
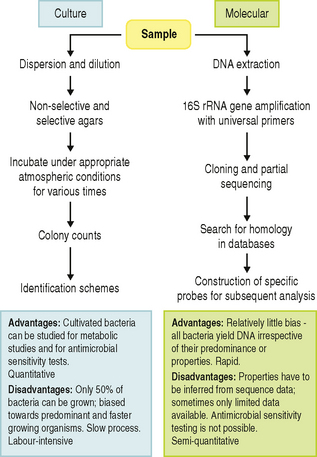
Classification is now dominated by comparisons of the sequence of 16S ribosomal RNA genes (16S rRNA). Within the rRNA gene, some stretches of DNA sequence are conserved while other areas are highly variable and reflect evolutionary divergence. These genes are around 1500 nucleotides long, which is short enough for rapid sequencing in conventional laboratories (using automated DNA sequencing equipment) but long enough to provide valuable discriminatory information to demonstrate similarities and differences among strains. The conserved regions can be used as a template for the design of ‘universal’ PCR (polymerase chain reaction) oligonucleotide primers that can be used to amplify the rest of the 16S rRNA gene, which can then be sequenced to identify differences in the variable regions. The sequences can be compared with those from other microorganisms, and to sequences in nucleotide databases, so that the relationship of an isolate to known species can be determined and evolutionary (phylogenetic) trees can be developed. The technique is relatively rapid, and has facilitated the analysis of a far wider range of bacteria than was previously possible. Comparison of 16S rRNA gene sequences have revolutionized the field of microbial taxonomy, and has clarified the classification of many previously heterogeneous groups of oral bacteria, such as the streptococci (Table 3.3) and anaerobic Gram positive rods formerly grouped as Eubacterium species (Fig. 3.5). In addition to classifying unknown strains, this approach can also be used to identify isolates, and has many advantages over conventional cultural approaches (see later; Fig. 3.2).
Table 3.3 Species of oral streptococci isolated from humans
| Group | Species | |
|---|---|---|
| mutans-group* | S. mutans | serotypes c,e, f, k |
| S. sobrinus | seroype d, g | |
| S. criceti | serotype a | |
| S. ratti | serotype b | |
| salivarius-group | S. salivarius | |
| S. vestibularis | ||
| anginosus-group | S. constellatus | |
| S. intermedius | ||
| S. anginosus | ||
| mitis-group | S. sanguinis | |
| S. gordonii | ||
| S. parasanguinis | ||
| S. oralis | ||
| S. mitis | ||
| S. cristatus | ||
| S. oligofermentans | ||
| S. sinensis | ||
| S. australis | ||
| S. peroris | ||
| S. infantis | ||
* mutans streptococci also include S. ferus (isolated from rats), S. macacae and S. downei (serotype h) (isolated from monkeys).
A consequence of classification is the proposal of new species. A species represents a collection of strains that share many features in common, and which differ considerably from other strains. Once a species has been recognised, then a type strain is nominated that has properties representative of the species. Type strains are held in national collections, such as the American Type Culture Collection, ATCC, or the National Collection of Type Cultures, NCTC, which is located in the United Kingdom.
A species may be divided into subspecies if minor but consistent phenotypic variations can be recognised. Likewise, groups of strains within a species can sometimes be distinguished by a special characteristic. For example, strains with a special biochemical or physiological property are termed biovars or biotypes, while strains with a distinctive antigenic composition are described as serovars or serotypes, and can be recognised using appropriate antibodies. Molecular approaches can also be adapted for subtyping strains within a species. Whole genomic DNA can be digested by different restriction enzymes (endonucleases), which cut the nucleic acid in specific places; these digests are then electrophoresed on an agarose gel to generate a chromosomal fingerprint. Different strains generate different patterns (restriction fragment length polymorphisms, RFLPs), although strains that appear to give similar patterns need to be compared after digestion with more than one enzyme. This approach can yield patterns too complex to analyse. In order to overcome this problem, the restriction fragments can be blotted onto nitrocellulose or nylon membranes, and hybridized with a suitably labelled probe to give a simpler profile. A ‘universal probe’ can be used, based on part of the rRNA gene sequence, and this can yield different patterns within a species (ribotyping). Ribotyping has allowed insights to be made into the transmission of strains among family members, especially from mothers to their babies, and in demonstrating the turnover of specific ribotypes (and specific clones) at a site (Ch. 4).
Principles of conventional microbial identification
Once organisms have been correctly classified using rigorous techniques, then more simple identification schemes can be devised in which only limited numbers of key discriminatory properties are compared (Fig. 3.1, Table 3.2). The first stage might involve the reaction of an organism with the Gram stain, and the determination of cellular morphology. Bacteria are then described as being, for example, Gram positive cocci or Gram negative rods, etc. Depending on the outcome of that division, simple physiological tests may be performed, such as the determination of sugar fermentation patterns, the profiles of acidic fermentation products following glucose metabolism, or selected enzyme activities. The rapid detection (ca. 4 hours) of constitutively-expressed enzymes by concentrated suspensions of bacteria has simplified the identification of some groups of bacteria. Ecologically-relevant substrates that detect enzymes such as glycosidases that cleave sugar residues from salivary mucins are now more commonly used to differentiate groups of bacteria that had previously been difficult to separate, for example, some oral streptococci. Some of these tests have been incorporated in kits and sold commercially, together with computerized databases, to facilitate identification.
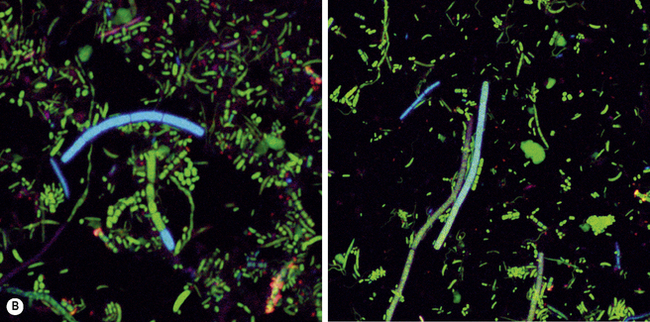
Monoclonal antibodies and nucleic acid (oligonucleotide) probes have been developed for the rapid identification of some species, but primarily those associated with disease. Such antibodies and probes can be labelled with a signalling group to aid in detection; examples include fluorescent dyes (Fig. 3.3), radiolabels, or enzymes such as horseradish peroxidase. These techniques have the advantage that organisms can be detected directly in plaque or a clinical sample without the need for lengthy culturing, although a potential drawback is that they can detect dead as well as viable cells.

Conventional microbial identification schemes can only be used when organisms have been isolated and grown in pure culture. Inevitably, the procedure for achieving pure culture (sample dispersion and dilution, growth on selective and non-selective agar plates, incubation conditions, etc) leads to the introduction of bias towards those microorganisms that grow quickly and easily under laboratory conditions (Fig. 3.2). Alternative, culture-independent, procedures have evolved out of modern taxonomic approaches, and these are giving a more accurate picture of the diversity (richness) of the microflora from a wide range of habitats.
The impact of molecular microbial ecology
As stated above, the genetic relatedness of microorganisms is now primarily determined by comparisons of 16S ribosomal RNA (rRNA) gene sequences. The biggest impact of this approach has been in the analysis of diverse communities of microorganisms from a number of habitats (molecular microbial ecology), including the mouth. Comparisons of the number of cells in samples that can be observed by microscopy versus those that can be cultured in the laboratory, even when using the most advanced techniques, has shown that only a proportion of the microflora at a site can be grown. The culturable fraction can vary from less than 1% of the total cell count in some marine habitats to about 50% of the oral microflora. These organisms that cannot currently be grown are termed ‘unculturable’; the reasons for this may be ignorance of an essential nutrient or other growth requirement, or because they evolved to grow as part of a community of cells, rather than as an isolated pure culture. Molecular ecological approaches have enabled the construction of phylogenetic trees that include currently unculturable organisms, so that in many cases, a genus and species name can still be assigned. Oligonucleotide probes can also be synthesized so that the presence of these organisms can be determined relatively simply in clinical samples using rapid tests, such as the polymerase chain reaction (PCR) or by in situ hybridization, usually with a fluorescent label (Fig. 3.3). An important benefit of these PCR-based molecular approaches is their potential to detect organisms that are present in low numbers. Although the properties of these uncultivable organisms cannot be determined using conventional tests (such as sugar fermentation patterns or their antibiotic sensitivity profile), databases of genes exist that can be interrogated to search for homologous sequences with known functions. This can provide insights into potentially important properties of these unculturable organisms, such as their cell wall structure, their virulence attributes, and the metabolic pathways they might use.
The existence of these unculturable bacteria cannot be ignored, since their presence at a site could be highly significant; for example, the aetiological agent of syphilis is a spirochaete, Treponema pallidum, that still cannot be grown in the laboratory. Two large families of novel unculturable bacteria have been identified in the mouth (Fig. 3.3), and these are commonly detected in deep periodontal pockets; further details will be presented later in this Chapter. In addition, some genera contain examples of both culturable and unculturable species; for example, there are around 50 Treponema species that can be seen by microscopy and detected by molecular approaches but which cannot be grown (Fig. 3.3A). Similarly, the molecular analysis of the microflora associated with dento-alveolar abscesses and endodontic infections has consistently identified novel groups of bacteria that were not recognised, or were grossly under-estimated, in parallel cultural studies (Fig. 3.2).
Difficulties arising from recent advances in microbial classification
Although recent advances have led to improvements in the classification of oral bacteria, such improvements have also generated a number of difficulties when interpreting or comparing early data when a previous (and sometimes flawed) nomenclature was in use. The classification of many groups of oral bacteria has been revolutionized in a relatively short time period, with many new genera and species described. A species highlighted in an early study may now have been reclassified and hence renamed, and so new and old terminologies may coexist in the scientific literature. For example, Streptococcus sanguis has been reported in the literature for many decades but, as of 1989, its description became more limited and organisms that were previously included within this species are now known to be sufficiently different as to warrant a distinct species epithet, e.g. S. gordonii. Consequently, some strains reported in earlier studies as S. sanguis may not have the same properties as strains more recently identified as S. sanguis sensu stricto. Furthermore, the Latin name originally given to some of the oral streptococci has recently been shown to be erroneous, and so S. sanguis is now termed S. sanguinis. For similar reasons, S. parasanguis, S. rattus, S. cricetus and S. crista have been renamed as S. parasanguinis, S. ratti, S. criceti and S. cristatus, respectively. Thus, great care has to be taken when interpreting older (and not so old) scientific literature.
Gram positive cocci
Streptococcus
Streptococci have been isolated from all sites in the mouth and comprise a large proportion of the resident cultivable oral microflora. The majority are alpha-haemolytic (partial haemolysis) on blood agar and early workers grouped them together, calling them viridans streptococci. However, haemolysis is not a reliable property in distinguishing these streptococci, and many oral species contain strains showing all 3 types of haemolysis (alpha, beta and gamma). These ‘viridans’-group streptococci are now clustered into four groups (Table 3.3) and will now be described.
Mutans-group (mutans streptococci)
There is great interest in the mutans streptococci because of their role in the aetiology of dental caries. S. mutans was originally isolated from carious human teeth by Clarke in 1924 and, shortly afterwards, was recovered from a case of infective endocarditis (growth of bacteria on damaged heart valves). Little attention was paid to this species until the 1960s when it was demonstrated that caries could be experimentally-induced and transmitted in animals artificially-infected with strains resembling S. mutans. The name of this species derives from the fact that cells can lose their coccal morphology and often appear as short rods or as cocco-bacilli (Fig. 3.4A). Nine serotypes have been recognised (a–h, and k), based on the serological specificity of carbohydrate antigens located in the cell wall, although some serotypes are found only in animals. Subsequent work showed that sufficient differences existed between cluste/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


