More Metals
Titanium and its alloys are of increasing interest in medical and dental applications because of their biocompatibility. However, these metals have properties that differ significantly from normal expectations, and recognition of this is important to their understanding. Key to this is the existence of allotropic forms and deformation twins.
Nickel-titanium goes further in having two very special behaviours: pseudoelasticity, shape-memory, dependent on a martensitic transformation. These allow a range of novel types of application.
Where casting is not appropriate, various other methods of forming metals are available, mostly mechanically. There are various advantages and disadvantages with these, depending on factors such as ductility and work-hardening. Superplastic forming, however, relies on a new mode of deformation: grain switching.
Pure gold for direct filling may be little used these days, and while relying on cold-welding, it can be understood in familiar terms: annealing and strain-hardening.
Dental mouth mirrors provide a nice case-study in the selection of materials according to several properties.
Mercury provides several challenges in its handling, storage and usage because of its toxicity, both directly to the user and environmentally. These matters are set out for reference. In this context, the obsolete restorative material copper amalgam is described fully in order to understand its demise. A related toxicity issue has to do with beryllium, a metal that should no longer be used in dentistry.
Dentistry employs many other other alloys for various purposes: Elgiloy is related to cobalt-chromium casting alloys, but also has features similar to titanium, as well as a further mechanism for hardening through a heat-treatment.
In addition to the common classes of metals and alloys that have already been covered, there remain some of more specialist use which have aspects of particular interest. It is the purpose of the present chapter to explore some of these systems. Again, it is broadly a matter of the relationship of properties and behaviour to the requirements of an application that control use, but in order to achieve the desired outcome recognition of the principles and limitations is necessary.
§1 Titanium
Titanium has long been known for its valuable combination of high strength with low density. Coupling this with remarkable corrosion resistance (it is unaffected by both acids and alkalis at room temperature), its application in diverse fields is not surprising,[1] and its use in medicine and dentistry has been assured for some time. It is used in various forms: commercially-pure metal (§1.1), α-alloys (§), and nickel-titanium (§2). It is a relatively expensive metal in part because of the need for special processing conditions using vacuum or an inert atmosphere; this arises from its extremely high reactivity with oxygen (a fracture surface ignites spontaneously in oxygen at 25 bar, and the molten metal just in air). However, it is this reactivity that leads, in the presence of atmospheric oxygen, to the immediate formation on the metal at ordinary temperatures of an oxide layer (TiO2) that is both adherent and stable, even in quite acidic conditions. This passivation (13§5) therefore inhibits further corrosion.
•1.1 Commercially-pure titanium
Titanium itself has two allotropic forms designated α and β. The α-phase has the h.c.p. structure (11§3.4) and is stable up to the transus temperature of 882 °C, at which it converts to the b.c.c. β-phase. This process is similar to the transformations occurring in iron (21§1). The α–phase is ductile, as is to be expected from the structure, but this is adversely affected markedly by dissolved (interstitial) oxygen (Fig. 1.1), and so the choice of grade of commercially-pure (CP) titanium used for a given application can be important.
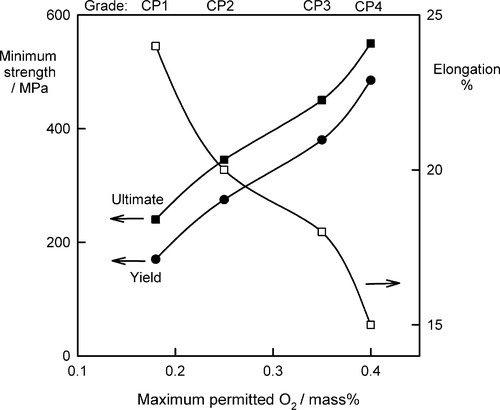
The effect of oxygen is so marked that it is commonly said that CP-Ti is better considered as an α-phase oxygen alloy. Carbon and nitrogen are also soluble interstitially and have similar effects on mechanical properties, such that one can write the oxygen equivalent thus:
< ?xml:namespace prefix = "mml" ns = "http://www.w3.org/1998/Math/MathML" />
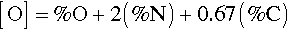
where each 0.1% O equivalent increases the strength by 100 ~ 120 MPa, but with a decrease in toughness (i.e. increase in brittleness).
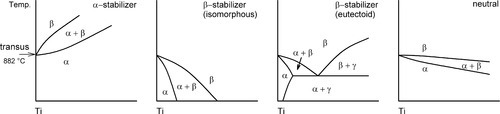
Many metallic elements are soluble to a greater or lesser extent in titanium, giving many alloying possibilities, although few of these have yet found applications in dentistry. Many of these additions have an effect on the transus temperature and accordingly are commonly classified as follows:
• α-stabilizers (e.g. Al, O, N, C)
• β-stabilizers, in turn divided into two groups:
– isomorphous (e.g. V, Mo, Nb, Ta, Re)
– eutectoid (e.g. Fe, Mn, Co, Cr, Ni, Si, H, Cu, Ag, Au)
• neutral (e.g. Zr, Hf, Sn)
An α-stabilizer raises the transus temperature, a β-stabilizer does the reverse, allowing the β phase to be stable at low temperature, but it may destabilize the solid solution such that it undergoes a eutectoid reaction (12§3.6). A neutral solute in principle has no effect on the transus, although in practice this is a matter of degree rather than an absolute statement, there being only relatively slight changes. These effects are shown schematically in Fig. 1.2.
The significance of the existence of the α-β transus lies in the behaviour of the cast metal: the initially-solidified metal is necessarily β-phase, because this is the stable phase at high temperature, and this must therefore transform on cooling. This transformation occurs in two stages, by a mechanism which is already familiar: nucleation and diffusional growth. Thus, at a temperature depending on the alloy and the rate of cooling, α-phase first nucleates at β-grain boundaries, producing a layer over all β-grains, but then further nucleation occurs allowing plate-like crystals of α to grow into the β-grains. Nucleation completely within β-grains can also occur, especially if the cooling rate is increased. This produces a Widmanstätten structure (Fig. 1.3) 1[2] (cf. Fig. 14§3.9), and eventually would, in pure Ti, be expected to result in a full conversion to α-phase. However, the existence of iron as a common contaminant, although of strictly controlled amounts in CP-Ti, as well as other elements, means that a small proportion of β-phase normally remains (some 2 ~ 5 vol% is typical), stabilized by the iron, even after annealing. This effect is enhanced by the partitioning of solute elements: β-stabilizing elements migrate into the remaining β-phase, while α-stabilizers become enriched in the α-phase, reinforcing the stability of the α + β two-phase system. In addition, the α-phase grain size is limited during the recrystallization of annealing by the presence of these remnants of the β-matrix. All this means that cast “pure” Ti is nothing like as ductile as otherwise would be expected: the fine grain, incoherent two-phase structure provides appreciable resistance to slip (cf. grain refining, 19§1.10). It also means that what is commonly described as “α” material is in fact more accurately interpreted as two-phase. Thus, although the alloying elements described above may not be used deliberately in ‘titanium’ as such in dentistry, the presence of any of them as impurities affects the behaviour of “pure” titanium. Clearly, it is of great importance to avoid contamination in casting titanium, much more so than perhaps with gold alloys, for example.

•1.2 Twinning
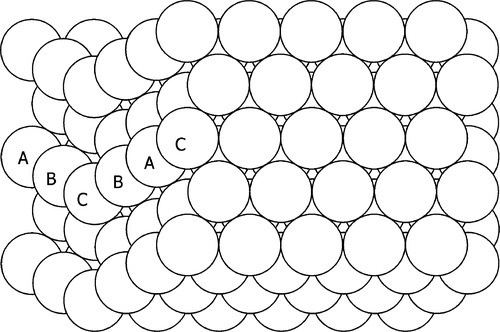
It will be recalled from 11§3.5 that the pattern of stacking of close-packed planes in the face-centred cubic (f.c.c.) structure may be characterized as A,B,C,A,B,C … , referring to the relative position of atoms in successive planes with respect to those in an (arbitrary) reference layer. As can be seen from Fig. 11§3.20, it can be visualized as a regular stepping displacement of each layer in a particular direction as a result of this. But, as is apparent from Fig. 11§3.17, the h.c.p. structure is obtained when this displacement occurs in alternating directions 60° apart for successive layers. Now consider just one alternation of this kind in the f.c.c. sequence, giving for example: A,B,C,A,B,C,B,A,C,B,A,C …. From the underlined “C” onward it is still the f.c.c. pattern, but in a different directional sense and so the letter sequence is inverted (Fig. 1.4). Notice that these ABC labelled atoms (Fig. 11§3.20) constitute the close-packed diagonal of the unit cell. Thus, if the sequence undergoes such a switch the diagonal direction must thereby be changed, and so the crystallographic axes themselves have a different orientation. The structure is crystallographically identical, and the boundary is perfectly coherent, but it is as if it has been reflected across a mirror plane. Such a crystal is said to be twinned.
Twinning can arise in two ways: firstly, accidentally during solidification or recrystallization, because the energy of this most perfect of grain boundaries is so very small that this kind of stacking error has little energetic penalty (and so little thermodynamic driving force to change); secondly, as a result of plastic deformation – giving deformation twins. For this latter case, it may be envisaged that from a normal f.c.c. crystal a twin may be generated by displacing a layer one position across, then the second, and the third, and so on. Since it is a shearing process it is also said to generate shear twins. It occurs in some metals and alloys as a normal result of applying tensile or compressive stresses – recall that slip occurs along close-packed planes even though these may be at some angle to the principal stress direction. It is worth re-emphasising that, despite the simplistic description given here, such slip is not homogeneous but must occur atom by atom (11§5,6). Nevertheless, the key point in such a process is that it is non-diffusive: every atom retains precisely the same set of nearest neighbours and they are said to move “in concert” (at least, as viewed after the event). In addition, although such rearrangements are driven by changes in lattice dimensions or orientations, because the crystal structure type is unchanged, there is no change in volume except for the accumulation of voids at grain boundaries as the deformation in adjacent grains cannot match exactly (Fig. 11§6.8).
Twinning can occur in several crystallographic systems, including types in which a crystallographic lattice rotation occurs as opposed to a reflection, although the f.c.c. example is perhaps the simplest. Thus, pure α-Ti – h.c.p. – readily shows deformation twinning. However, this mode of response to applied stress is suppressed by the presence of solute atoms such as aluminium and oxygen (α-stabilizers), and so only occurs in low-oxygen CP-Ti. Even so, the ductility of very pure α-Ti (if it were obtained) would be due to this response in addition to the more usual slip by the movement of dislocations (11§6). Single-phase β-Ti alloys (b.c.c.) also exhibit deformation twinning, a behaviour which in fact enhances the ductility of what is ordinarily understood to be a relatively brittle crystal structure. In addition, interstitial oxygen does not cause the great lattice distortion that it does in b.c.c. iron and steel (metallic radius is much larger – Ti: 147 pm, Fe: 126), so slip remains relatively much easier even in the presence of this common contaminant.
Thus we have the curious situation that in practice α-Ti is expected to be ductile but is not because of an actual two-phase structure, grain boundary effects and distortion by interstitial contaminants; on the other hand, β-Ti is expected to be relatively brittle but is instead ductile because of twinning and lack of effect of interstitials.
•1.3 Casting
As indicated above, titanium is extremely reactive and great precautions must be taken in the process of casting the metal. In part this is due to the high m.p., 1670 °C, substantially above that of Co-Cr alloys, and special equipment is therefore necessary. In addition, this must provide an inert atmosphere, such as with argon or vacuum. Even then, care must be taken to exclude oxygen after casting because above ~ 550 °C oxygen diffusion through any oxide coating and into the metal is very fast, leading to severe embrittlement as well as an excessively thick oxide coating. The embrittlement arises because the solid solubility of oxygen in Ti is very high (~ 14.5 mass% = 33 at%) and forms an oxygen-diffusion zone beneath the actual oxide layer. This “α-case” forms even on β-alloys, and may be a source of cracking in tension even if the underlying metal is ductile. The oxide commonly appears dark blue or other colours, partly through interference effects, but also because it is anion-deficient, creating colour centres (cf. ZnO, 9§3.1). This anion-deficiency is the source of the high diffusion rate of oxygen through the oxide – there are vacancies at the oxygen sites.
Cooling of the casting well below that critical temperature of ~ 550 °C must therefore be allowed to occur in the same inert atmosphere to avoid these effects. A further reaction problem arises with the investment if this contains silica, as indicated earlier (17§4): titanium can reduce silica to silicon.
The low density of the pure metal (4.5 g/mL) also causes difficulties. Centrifugal casting machines (18§3.2) are impractical because of the controlled-atmosphere requirements and gas-pressure (18§3.5) is commonly used in Ti-casting machines. Even so, turbulence porosity is often a major problem that requires careful control of spruing and casting conditions, especially in thicker sections. A further difficulty arises from the effects of other elements on the mechanical properties, as described above. Accordingly, casting with titanium must be done under scrupulously clean conditions to avoid contamination by other metals.
•1.4 β-Titanium
Elements that stabilize the β-structure have a curious effect: they can reduce the modulus of elasticity markedly when present in modest concentrations, to perhaps to 60 or 70% of the value for α-Ti at around room temperature (that is, from about 105 GPa). This is curious because ordinarily modulus of elasticity is little affected by anything other than temperature (assuming no constitutional changes) because it is a measure of interatomic forces – bond strength – and in chemical terms the transition elements involved are not that dissimilar. Thus it seems that β-stabilizing elements disturb that bonding. This effect is seen in such alloys as Ti-10Mo (meaning 10 mass% molybdenum), where there is enough molybdenum to stabilize the β-structure at around room temperature, and likewise for alloys near Ti-15 V-3Cr and a number of others. Such bond-weakening might also represent a further contribution to the ductility of this type of alloy, although heat-treatment after cold-work is likely to result in detrimental metallographic changes.
Some β-phase alloys are not fully stable at low (normal working) temperatures and can tend to produce the so-called ω-phase, a very fine grain (2 ~ 4 nm) precipitate, yet again by a diffusionless shear-transformation, as a hexagonal or distorted b.c.c. structure which has a diffusely-coherent grain boundary with the β. This is not itself an equilibrium phase, but forms on nucleation in the transition from α to β. Such β-alloys are said to be metastable, and the presence of ω-phase causes severe embrittlement. Thus, heating of such alloys may be detrimental to their structure and thus their properties. Conditions of use must be carefully observed.
In dentistry, a particular titanium-molybdenum alloy, Ti-11Mo-7Zr-4Sn, with the trade name TMA, is used for orthodontic wires. This has the β-Ti structure, principally stabilized by the molybdenum content, but the presence of the zirconium and tin (otherwise nearly neutral with respect to the transus temperature, §1.1, but see §1.6) inhibits the formation of the brittle ω-phase. It is therefore weldable without detriment. The lack of Ni is an advantage for intraoral use.
•1.5 Corrosion
Although titanium and its alloys are corrosion-resistant, as with all passive alloys they are susceptible to crevice corrosion attack and all the normal remarks apply (13§5). However, there is an additional factor for CP-Ti and α-phase alloys, especially with aluminium, and that is hydrogen embrittlement. This generally arises because the cathode process
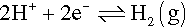
(13§3) can occur readily enough to cause a problem: hydrogen is sufficiently soluble in α-Ti that the mechanical properties can be affected, in particular a loss of ductility, but it can be driven to supersaturation when the brittle phase TiH2 may form by precipitation. This precipitation is aided by the presence of tensile stress (reducing the constraint on the hydride’s crystal growth), and so stress-concentration by notches becomes a factor of relevance in focusing the embrittlement on just the region when crack growth needs to be suppressed by crack-tip blunting, i.e. where ductility would provide toughness.
The type of effect known as internal hydrogen embrittlement only occurs when hydrogen is present over and dissolves in the molten metal, which therefore becomes supersaturated with hydrogen on solidification. This is not normally likely to occur in ordinary dental laboratory contexts but might be a problem at manufacture. Environmental hydrogen embrittlement, on the other hand, results from hydrogen absorption by solid metal. In dentistry, this might occur during electrolytic processes or, more likely, from corrosion reactions. This seems to be the case in the presence of fluoride, and can result in changes in the properties of α-phase orthodontic wires. Since these are meant to be stressed in use, delayed fracture (static fatigue) may occur in service.[3] While β-phase wires do absorb hydrogen, this stays in interstitial solution and may have only relatively minor mechanical effects.[4] Similarly, etching of castings, for example with strong sulphuric acid, whether for obtaining a mechanical key for cementation or to improve the biocompatibility of the surface of an implant, may generate much TiH2.
• 1.6 Zirconium & Hafnium
Both zirconium and hafnium are isomorphous with titanium, and show the same α-β transition, the temperature of which is only slightly affected in their alloys (hence their description as ‘neutral’) (Fig. 1.5). The metallic radii are Zr: 160 pm and Hf: 159 pm, giving radius ratios with Ti (147 pm), of 1.09 and 1.08 respectively. By the Hume-Rothery rules (Table 11§3.2) the complete solid-solubility that is observed is expected. In addition, both Zr and Hf are spontaneously passive (13§5), and the ability to form the coherent oxide coat, as has Ti, is present in the alloys. The radius ratios are still sufficient to generate appreciable solid solution hardening without employing alloying elements that pose a biological risk, such as Al, Ni, and Cr. The only significant problem is the high price of both metals. Commercially-pure grades are used for surgical implants.
§2 Nickel-Titanium
Nickel-titanium alloys have two main areas of application in dentistry, but with a third a distinct possibility. These areas are in orthodontic appliances, endodontic instruments, and surgical correction of bony anatomy. Each depends on properties that are commonly only encountered with this group of alloys (although others exist outside dentistry), but to understand these it is first necessary to explore some of the underlying structure.
•2.1 Transformations
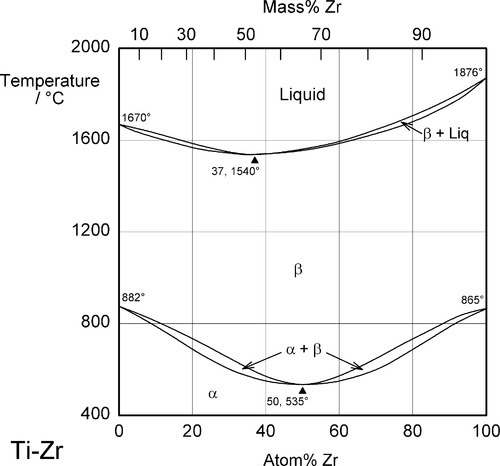
Nickel and titanium form a series of intermetallic compounds: Ni3Ti, NiTi and NiTi2. It is NiTi that is of relevance here, the equimolar alloy, or nearly so. But far from being a sharply defined ‘compound’ as are both Ni3Ti and NiTi2, it can be seen from the phase diagram (Fig. 2.1) that the compositional limits for NiTi can be quite broad above 630 °C in the eutectoid phase field. Notice that below this temperature there is a two-phase field that stretches from about 25 to 66% Ti, Ni3Ti + NiTi2. However, cooling NiTi itself below that temperature does not result in that phase separation, but the preservation of NiTi as a metastable phase.[5] Cold working aids the reaction.
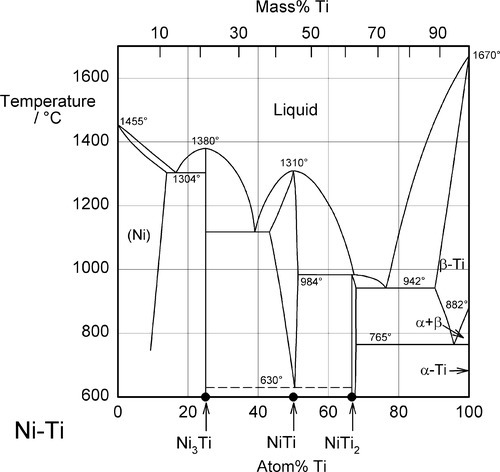
Also known as Ni-Ti (“nye-tye”) or “Nitinol” (from the US Naval Ordnance Laboratory where it was first investigated), nickel-titanium alloys approximating the equimolar composition NiTi are remarkable for exhibiting a range of unusual properties, in particular the shape-memory effect and pseudoelasticity. These arise from certain reversible crystallographic changes.
NiTi at relatively high temperatures has an ordered b.c.c. structure which is known in this system as austenite. On cooling, one observes the expected thermal contraction, more or less linearly, until at a particular temperature the rate of change of dimension with temperature increases. This is caused by a spontaneous but progressive (i.e. with respect to temperature, not time) shear transformation to a monoclinic structure called martensite.2 Such a change is therefore called a martensitic transformation, simply because crystallographic shear is involved. Hence, the temperature at which this process commences on cooling is called the martensitic start temperature, MS (Fig. 2.2). At some lower temperature, this process is completed: this is the martensitic finish temperature, Mf, where the proportion of martensite is now 100%. Below Mf, ordinary cooling shrinkage occurs as before. Notice that in this process, all neighbours are preserved (no diffusion), and therefore the monoclinic structure is also ordered.
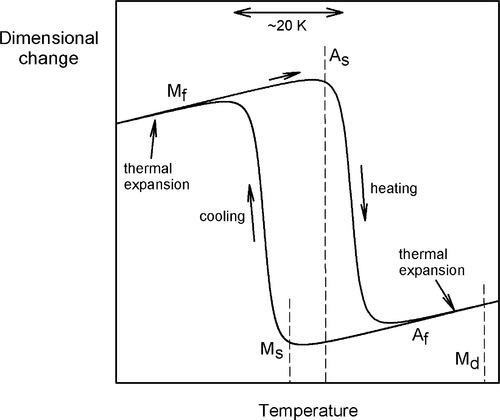
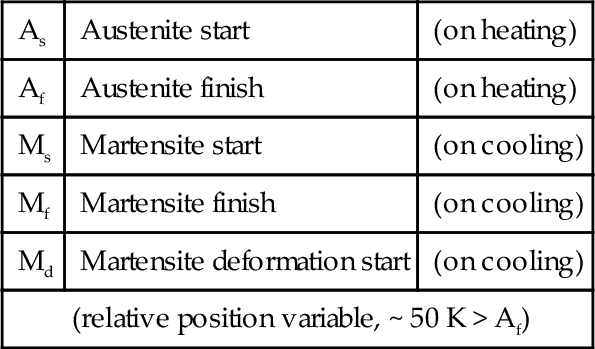
If now this martensitic NiTi is reheated, the reverse process occurs, eventually completely reforming the original austenite. That is to say, the shear transformation is progressively reversed so that all atoms return to their original positions in their original crystallographic arrangement. However, the austenitic start temperature, AS, is somewhat higher (typically by about 20 K) than Mf, and likewise the austenitic finish temperature, Af, is somewhat higher than MS. These transformations therefore show hysteresis, which occurs over the transformation temperature range (“TTR”). This whole process is more or less indefinitely reversible. Such a thermally-controlled crystallographic change is a normal consequence of the thermodynamic imperative of adopting a lower energy state, there being a simple path to allow it to occur and no limitation arising from diffusion. What is peculiar is the existence of the hysteresis; it is a second-order transition, unlike the first-order transition of ferrite to austenite and vice versa for iron (Fig. 21§1.1), for example. Thus, the change is not microscopically-reversible, as would be expected for a normal equilibrium process.
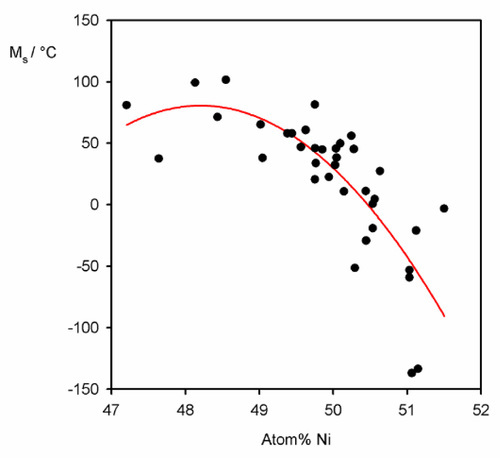
The progressive nature of the transformation may in part be explained like this. It is clear that if the composition departs from exactly 50:50 Ni:Ti in atomic proportions, it is not possible to maintain complete ordering over a whole grain (assuming that a second phase does not appear). However, this disorder will itself be randomly distributed. The energy conditions required for the transformation therefore vary slightly from place to place according to statistical fluctuations in the degree of disorder, and hence the temperature at which it occurs will also vary over some range. Further, even at exactly NiTi, stacking faults may occur in which Ni and Ti occupy the wrong sites, a further source of slight disorder affecting the strain energy of the lattice. The effect that this has can be seen by the variation of the value of MS with composition (Fig. 2.3). In addition, remembering that grains will have randomly-oriented crystallographic axes, the transformation will be slightly constrained by the stress generated as neighbouring regions and grains cannot exactly accommodate the strain. The driving-force needed therefore continues to increase slightly to overcome this resistance.
An extra complication to this behaviour is that some NiTi alloys also show a third phase, the so-called R-phase, after its rhombohedral crystal structure (Table 11§3.1). Generally, this only appears on cooling as an intermediate stage before the full martensitic transformation. Again, however, the formation of this involves only shear and so does not interfere with, but rather becomes part of the continuous process of transformation. We may ignore such details in favour of the larger picture.
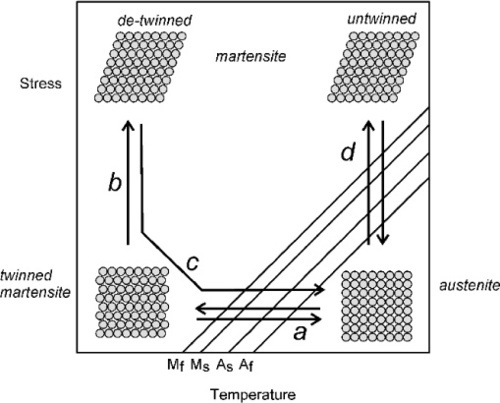
Austenitic NiTi shows quite ordinary mechanical behaviour at temperatures well above Af and has no special attributes; it is quite hard, and resembles α-Ti (§1.1). However, the martensite that is reversibly formed thermally, i.e. only on cooling, is heavily twinned (§1.2) in a special way: it may be imagined as similar to a pleated structure or a “concertina-fold” (Fig. 2.4, process a). It may be seen that the formation of this type of structure leads to a small change in volume (Fig. 2.2), but not of shape (we assume here that this is in a polycrystalline piece of metal, where the net strain is isotropic; cf. Fig. 1§2.12) as there is no systematic displacement from layer to layer. This twinned martensite does have special properties: when this is mechanically deformed, the twinning is progressively lost until it has completely disappeared (Fig. 2.4, process b), again through a plain shearing mechanism, with all near neighbours preserved. Again, this is a regimented (or ‘military’) non-diffusive crystallographic change. In this way, very large strains can be obtained but at nearly constant, only very slightly increasing load. Furthermore, this strain too is not microscopically-reversible because it is not an elastic strain: atoms have moved from one energy minimum to another through, of necessity, a (slightly) higher energy state. This is quite unlike the elasticity arising from bond-length or bond-angle strain alone; removing the stress does not result in the strain returning promptly to zero, only the purely elastic component of the deformation is recovered. The nearly-atomic scale of shifts occurring mean that strains up to about 8% may be obtained. This should be compared with the 0.1 ~ 0.2% strain at the elastic limit of many other alloys, which is therefore about the magnitude of the pure elastic component here.
•2.2 Shape-memory
The deformed, de-twinned martensite is thus stable in the absence of any applied stress if kept at low temperature, relative that is to AS. However, if it is now heated to above AS it becomes unstable and spontaneously reverts to austenite (Fig. 2.4, process c). Yet again, because this is purely a shear transformation, all neighbours are still preserved. Consequently, the original dimensions of the piece are regained because all of the imposed strain is recovered. Under this regime, therefore, NiTi demonstrates shape-memory, and is therefore called a shape memory alloy (“SMA”), a property it shares with a variety of other alloys that are not used in dentistry. It is clear that work has been done in creating the deformed martensite with respect to the original austenite, and this work is stored in the structure. Hence, when heated, this work is recoverable and can be used to exert forces and thus in turn do work on an external system. It is similar to the work done by the energy stored (purely elastically) in an ordinary spring, such as in driving a clockwork mechanism. The difference here is that the release in NiTi is triggered by the imposed rise in temperature. Obviously, the deformation can be of any type: in tension, bending, or torsion.
The low-temperature martensitic phase is very plastic. A piece can be deformed in a manner rather similar to that of pure tin: it can repeatedly be bent, backwards and forwards, without strain hardening. This enables any necessary shaping to be done with little risk of fracture, unlike metals such as stainless steel and dental gold alloys. The reason for this is that the activation energy for the movement of the twin boundaries is low, and – again unlike other alloys – without the formation or involvement of dislocations, which through pile-up would otherwise lead to the formation of microscopic defects that can initiate fracture (11§6.3). The remarkable bonus is that the original shape can then be recovered just by mild heating.
For obvious reasons, the above shape-memory behaviour is called “one-way”: a mechanical deformation must be imposed first, and this is undone on heating above AS. The initial thermally-induced transition is, overall, dimensionally isotropic because the b.c.c. austenite crystal axis that becomes the c-axis in the monoclinic martensite is chosen randomly from grain to grain (cf. the formation of the ordered AuCu domains, 19§1.4). However, it is possible through elaborate combinations of thermal and mechanical treatments to make the martensitic transformation on cooling anisotropic, that is, obtain a selective orientation of the martensite c-axis. Thus, cooling through that transformation also causes a dimensional change without loading, and this is recoverable on reheating. This “two-way” shape-memory can therefore do useful work in either direction. Note that the thermal hysteresis is still present, although it may be reduced in extent. The details of such shape training are, however, beyond the present scope.
•2.3 Pseudoelasticity
There is a third special behaviour. Austenitic NiTi is said to be mechanically unstable over a narrow range of temperatures. That is, in this temperature range the martensitic transformation can be induced purely mechanically, by the imposition of a stress. This temperature range is marked above by what is termed the martensite deformation start temperature, Md (typically up to about 50 K above Af), and below by Mf, where there is no austenite remaining. For the moment, however, we shall restrict the discussion to temperatures above Af, that is, where no martensite is present. Thus, for a piece of austenitic NiTi in that temperature range, the application of a load first causes a perfectly ordinary elastic deformation with an unremarkable modulus of elasticity (Fig. 2.5). However, at some point the stress-induced shear transformation commences and, if the load is increased slightly, more austenite transforms to untwinned martensite, and so on until all has been converted. The transformation is again progressive and second-order. Very large strains, again of the order of 8%, can thereby be obtained with only very small increases in stress. This is the phenomenon of superelasticity, also known as pseudoelasticity, which is perhaps the better, more accurately descriptive label. After the completion of the conversion the elasticity again becomes normal – the metal is stiff and elastic – until an ordinary yield point is reached. Remarkably, if the load is reduced before that (true) yield has occurred, the mechanical deformation is reversible, both the ordinary elasticity (of course) and the pseudoelastic deformation due to the martensitic transformation. This is represented in Fig. 2.4 by process d. It necessarily involves the direct formation of the untwinned martensite because of the imposed strain, and the structure is indistinguishable from that formed by stress at low temperature (except for ordinary thermal expansion). Lowering the temperature while under stress has no effect on structure. Again, there is hysteresis: the unloading curve sees the start and finish of the return to austenite at lower stresses than caused the martensite to form (Fig. 2.5); the loading and unloading curves in this region do not coincide.
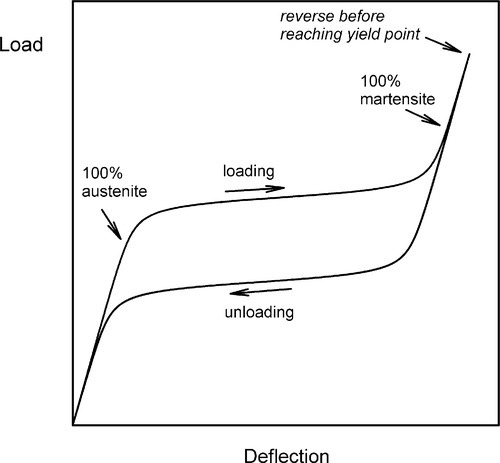
As might be imagined, there is some temperature- and composition-sensitivity to the value of the stress that induces the transformation. In fact, Md represents the highest temperature at which this may be achieved and it also requires the highest stress to initiate. This arises because above that point the critical transformation stress becomes inaccessible, being higher than the yield point for ordinary, irreversible plastic deformation of the austenite (that is, by the movement of dislocations), which is therefore the path taken, according to the principle of least work. At lower temperatures, the martensite becomes energetically progressively more favourable and the activation energy for the transformation, as represented by the stored (true) elastic energy, lower.
Although the stress to induce the transformation is temperature-dependent, the speed of the transformation is dependent on neither temperature nor strain rate. It is indeed a fast transformation, occurring at about half the speed of sound in the metal (that is, at about 2500 m/s), in other words, at half the speed of transmission of the elastic wave. Thus, for small pieces, the progressive transformation occurs essentially instantaneously. Similarly, the temperature-induced transformations are equally fast, and do not require any more time to complete. All of this arises from the non-diffusive nature of the crystallographic changes.
Now, if instead of starting at T > Af, the loading occurs for Af > T > AS, the material is, of course, 100% austenite to start with, and can stress-transform />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


