Hemifacial Microsomia
Evaluation and Treatment
• Considerations during Infancy and into Early Childhood
• Dysmorphology Associated with Hemifacial Microsomia
• Facial Growth Potential with Hemifacial Microsomia
• Classification of Temporomandibular Joint–Mandibular Malformation
• Staging of Skeletal Reconstruction: Timing and Techniques
• Consideration of First-Stage Mandibular Reconstruction during the Mixed Dentition
• Facial Soft-Tissue Reconstruction
Hemifacial microsomia (HFM) is a craniofacial malformation that results in varying degrees of hypoplasia of the structures within the first and second branchial archs.34,35,38,53,62,65,66,81,84,174,227 Congenital hypoplasia is generally unilateral, although bilateral (asymmetric) involvement occurs in 5% to 15% of patients. Most cases of this condition are sporadic, but there are rare familial causes that exhibit autosomal dominant inheritance. The term hemifacial microsomia was first used by Gorlin and Pindborg during the early 1960s, although the first recorded cases may have been those of Canton in 1861 and Von Arlt in 1881.62 Many terms have been used for this malformation, thus indicating the wide spectrum of anomalies observed and emphasized by authors from various disciplines. In addition to HFM, the malformation has been called craniofacial microsomia, oculo–auriculo–vertebral dysplasia, first and second branchial arch syndrome, lateral facial dysplasia, unilateral oto–mandibular dysostosis, facio–auriculo–vertebral sequence, and oculo–auriculo–vertebral spectrum.
Inheritance Pattern
The occurrence of HFM has been estimated to range from 1 in 3500 to 1 in 26,550 live births. Cohen suggests that the birth prevalence is likely to be around 1 in 5600 live births.62,26 Morrison and colleagues stated a prevalence rate of conditions on the oculo–auriculo–vertebral spectrum of 1 in 45,000 in Northern Ireland.26
Kelberman and colleagues performed a genome-wide search for linkage in two families with features of hemifacial microsomia.105 In one of these families, the data were highly suggestive of linkage to a region of approximately 10.7 cM on chromosome 14q32, with a maximum multipoint LOD score of 3.00 between microsatellite markers D14S987 and D14S65. Linkage to this region was excluded in the second family, thereby suggesting genetic heterogeneity. With the use of an animal model.
Poswillo demonstrated that early vascular disruption with expanding hematoma formation in utero resulted in the destruction of differentiating tissues in the region of the ear and the jaw.214–218 The degree of local destruction appeared to be related to the severity of tissue damage caused by the hematoma. The constellation of anomalies seen in patients with HFM suggests an origin that occurs at about 30 to 45 days’ gestation. Naora and colleagues described a transgenic mouse line that carried an autosomal dominant insertion mutation that resulted in facial anomalies that resemble HFM, including microtia and abnormal occlusion.164
Engiz and colleagues reviewed the clinical and laboratory findings of 31 individuals with Goldenhar syndrome (15 boys and 16 girls) between the ages of 1 day and 16 years.47 The characteristic features were preauricular skin tags (90%), microtia (52%), hemifacial microsomia (77%), and epibulbar dermoids (39%). Vertebral anomalies were noted in 70%, cardiac malformations were found in 39%, genitourinary anomaly were noted in 23%, and various central nervous system malformations were found in 47%.
Several classification systems for HFM have focused on one or more fundamental anatomic features of this anomaly. In 1991, Vento and colleagues described the OMENS classification for HFM. This system substratifies each of five anatomic manifestations of HFM in accordance with their dysmorphic severity on a scale from 0 to 3. The five manifestations each constitute one letter of the OMENS acronym: orbital asymmetry; mandibular hypoplasia; ear deformity; nerve dysfunction; and soft-tissue deficiency. Scoring was done on the basis of conventional radiographs (e.g., posteroanterior, lateral, submental, panoramic), physical examinations, and photographs. Although the OMENS classification does allow for the objective cataloguing of a range of the abnormalities that constitute the spectrum of HFM, it falls short in terms of providing effective communication when determining the timing and technique that are best suited for the reconstruction of each component of the anomaly (i.e., maxillo-mandibular, orbito–zygomatic, soft tissues, external ear). It also is of limited value with regard to overall patient management.276
At the present time, HFM should be regarded as a nonspecific symptom complex that is etiologically and pathogenetically heterogeneous.* Extreme variability of expression is the characteristic finding.
Considerations during Infancy and into Early Childhood
At the time of birth, for a child with HFM, concerns will center on the adequacy of the airway; swallowing and feeding; hearing; vision; the presence of other associated malformations; and family unit psychosocial issues.3,16,27,28,56,57,112,125,131,165,175,193,196,197,199,207,208
The airway may be compromised as a result of 1) maxillary hypoplasia with choanal (nasal) obstruction 2) mandibular micrognathia with a retropositioned tongue obstructing the oropharyngeal/hypopharyngeal spaces 3) laryngomalacia/tracheomalacia or 4) neuromotor involvement.3,16,27,28,57,195 Depending on the severity of these malformations, a spectrum of airway problems may be present and necessitate treatment that ranges from special infant positioning with an extended hospital stay to mandibular osteotomies with advancement; on rare occasions, immediate or delayed tracheostomy may be required.
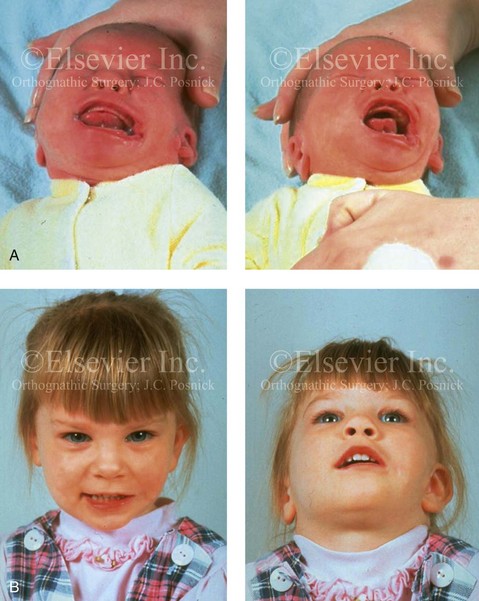
When a cleft (of the lip, palate, or both) or macrostomia is present, the timing of correction of these anomalies is generally similar to that used for the patient with non-syndromic cleft lip and palate (Fig. 28-1).
Dysmorphology Associated with Hemifacial Microsomia
Facial Soft Tissues
From a clinical perspective, the soft-tissue deficiencies associated with HFM can be considered to affect four anatomic regions of the head and neck: 1) the external ear; 2) the eyelid–adnexal structures; 3) the preauricular–cheek–lip soft tissues; and 4) the temporal fossa (Figs. 28-2 and 28-3).19,51,85,98,101,106,130,133,129,228,251 The soft tissues within each region that may be deficient and dysmorphic include the bulk of the cutaneous and subcutaneous tissue, the volume of fat, the muscles of mastication and facial expression, the cranial nerves, and the parotid and submandibular glands.
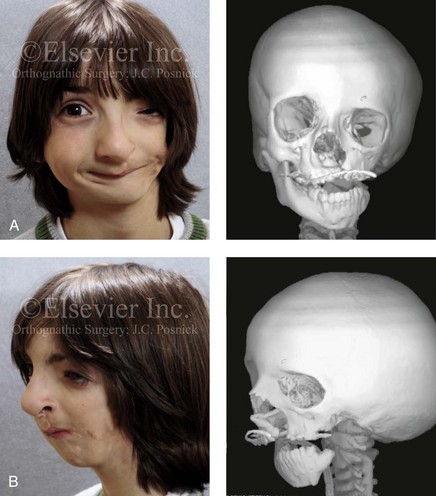
Figure 28-2 A 9-year-old boy who was born with left-sided hemifacial microsomia and a unilateral cleft lip and palate. He underwent lip and palate repair during early childhood at another institution. He is congenitally missing the left external ear and external auditory canal. The soft tissues of the eyelids and the cheek region are also deficient. The skeletal malformation involves the upper facial skeleton (i.e., the left zygomatic complex, the left orbit, and the left squamous temporal bone) and the lower facial skeleton (i.e., the maxilla and the mandible). He has a type III glenoid fossa mandibular malformation. A, Frontal facial and computed tomography scan views. B, Oblique facial and computed tomography scan views.
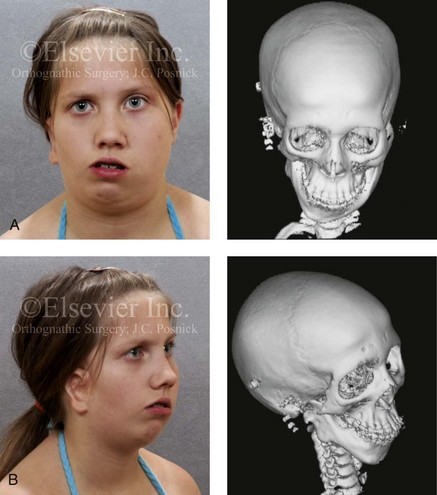
Figure 28-3 A 14-year-old girl who was born with right-sided hemifacial microsomia, including microtia and an absent external auditory canal. There is soft-tissue deficiency on the right side of the face. The skeletal malformations involve the right zygomatic complex as well as the mandible and the maxilla. There is a Type IIB mandibular malformation. The patient had previously undergone right ear reconstruction and the placement of a bone-anchored hearing aid (BAHA) at another institution. A, Frontal facial and computed tomography scan views. B, Oblique facial and computed tomography scan views.
According to the research of Kane and colleagues, in patients with HFM, the extent of hypoplasia of specific muscles of mastication frequently predicts the extent of dysplasia of the osseous origin and insertion of those muscles.98 If the temporalis muscle is hypoplastic, a deficiency of the coronoid process will be present. When the masseter muscle is hypoplastic, the gonial region of the mandible will also be deficient. When the lateral pterygoid muscle is deficient, the condylar head is deficient or absent.
Abnormalities of the external ear are a consistent finding and range from anotia to a mildly dysmorphic ear. Farkas and James were unable to demonstrate a direct relationship between the degree of microtia and the extent of skeletal deformity.51
The External Auditory Canal, the Middle- and Inner-Ear Structures, and Audiologic Findings
A narrow external auditory canal is frequently found in patients with mild external ear deformity, whereas atretic canals are expected in more severe cases.17,18,223,280 At times, a small external ear with normal middle-ear architecture is seen. Audiometry will delineate the nature of the hearing loss, which likely includes conductive and, less frequently, sensorineural loss in 15% of patients. Hypoplasia or agenesis of the ossicles may occur. In a comprehensive study, Caldarelli and colleagues used air and bone conduction audiometry and temporal bone tomography to evaluate 57 patients with hemifacial microsomia.17,18 The authors were unable to correlate the degree of auricular (i.e., external ear) deformity with hearing function. Focused temporal bone CT scans offer the best documentation of the middle-ear structures. Assessment of the unaffected and presumed normal ear is an essential part of the evaluation.
Maxillomandibular Region
A variable degree of hypoplasia of the skeletal structures within the first and second branchial arches will occur. As a result, the anteroposterior, transverse, and vertical dimensions of the face are diminished on the affected side, with secondary deformities on the contralateral side. This is especially true in the maxillomandibular region (see Figs. 28-2 and 28-3).134
Facial Growth Potential with Hemifacial Microsomia
The potential for longitudinal facial growth in a child who is born with HFM is an important factor when considering the timing and techniques of reconstruction.96,97,114,115,133,144–147,152–154,168,194,202–204,225,226,230–232 An additional pivotal consideration for the maxillofacial reconstruction of the individual with HFM is the integrity of the glenoid fossa–condyle–ascending ramus of the mandible (Fig. 28-4). Some authors speculate that a reduced mandibular growth potential secondarily leads to the observed maxillary deficiency on the ipsilateral side with progressive canting of the occlusal plane. Those who advocate mandibular surgery during the mixed dentition often do so with the belief that it will effectively correct the lower jaw malformation and that normal ongoing mandibular growth will continue with the prevention of secondary maxillary growth deformities in an otherwise normal upper jaw. There are objective clinical and radiographic studies in the literature that review the issue of the progression of deformity with HFM during growth.87–89,194,225,226
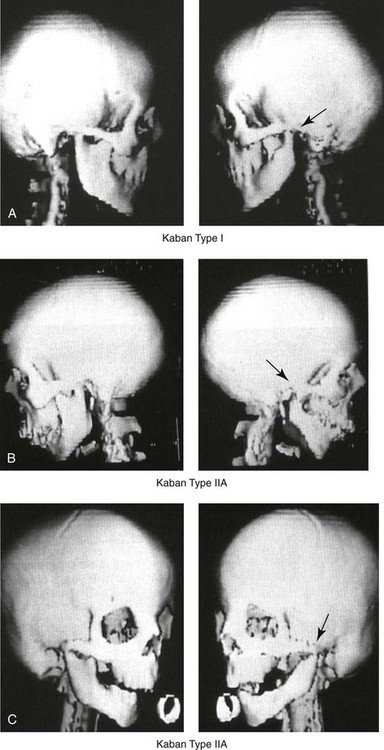
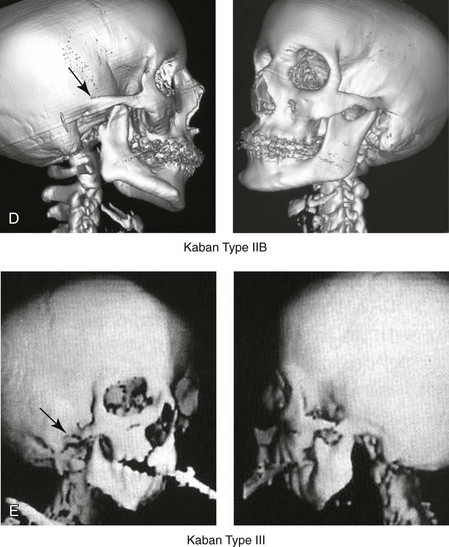
Figure 28-4 Kaban and colleagues described a classification system to define the degree of glenoid fossa–mandibular malformation observed in patients with hemifacial microsomia. Three-dimensional craniofacial computed tomography (CT) scans illustrate this classification system (see text for complete description). A, CT scans of a Type I mandibular malformation. B and C, CT scans of a Type IIA mandibular malformation. D, CT scans of a Type IIB mandibular malformation. E, CT scans of a Type III glenoid fossa–mandibular malformation in a patient who also has a unilateral cleft lip and palate.
Rune and associates placed metallic implants in the jaws to study the facial growth of 11 patients with HFM.225,226 They collected and studied serial roentgen stereophotogrammetry images from each patient. They found that, in five patients, the cant of the occlusal plane seemed to “slightly” increase; in the remaining six patients, the occlusal plane asymmetry either remained the same or improved with time. The authors also stated that the pattern of mandibular displacement showed no correlation with the severity of the presenting deformity. They concluded that the data “do not support the claim that the asymmetry of the jaws is invariably increased in time because of growth disparity between the affected and the unaffected sides.”225,226
Polley and associates completed a longitudinal radiographic cephalometric study of the maxillofacial region in patients with HFM.194 Twenty-six patients were included in the study and were followed longitudinally. Five patients (19%) had a Pruzansky grade I mandibular deformity, 14 patients (54%) had a Pruzansky grade II deformity, and 7 patient (27%) had a Pruzansky grade III deformity. The average age at the time that the initial cephalometric records for all patients were obtained was 3.5 years (range, 0.7 to 9.2 years), and the average age at the time that the final cephalometric records were obtained was 16.7 years (range, 10.1 to 22.5 years). None of the study patients had undergone surgical or orthopedic jaw manipulation during the time of the study, although nine patients underwent fixed appliance orthodontic treatment. Vertical and horizontal skeletal mandibular asymmetry was evaluated with posteroanterior cephalometry. The authors found that the growth of the affected side in patients with HFM parallels that of the contralateral side. The degree of mandibular asymmetry in the study patients remained relatively constant throughout craniofacial development. The mandibular skeletal deformity as measured in the study subjects did not progress with time. The growth rate of the affected side was similar to that of the contralateral side in each subject, irrespective of the degree of presenting mandibular deformity on the involved side. In the study group, subsequent growth occurred in both mandibular rami in each patient. This was true for all patients, irrespective of the Pruzansky grade and the side of the mandible that was affected.
Ongkosuwito and colleagues studied mandibular growth in 84 consecutive patients with a confirmed diagnosis of unilateral HFM. The mandibular malformation for each subject was categorized into one of four grades on the basis of the Pruzansky/Kaban classification. The malformations were then regrouped to reflect those with a functional glenoid fossa and condyle (Type I and Type IIA) and those without (Type IIB and Type III). The study groups were compared with a normal age-matched Danish control group. For each subject, an orthopantomogram was obtained and used to perform measurements of ramal height (i.e., the distance measured in millimeters between the condylion and the gonion) on each side. The data confirmed that patients with HFM start with a shorter ramal height and end up with a shorter ramal height, although growth is the same in both patients with HFM (regardless of the extent of malformation) and controls. In patients with HFM, the ramal height is smaller on both sides, which means that growth is characterized not simply by unilateral underdevelopment but by a complex three-dimensional phenomenon.161A
According to the independent findings of Polley and colleagues, Rune and colleagues, Meazzini and colleagues, Ongkosuwito and colleagues, the mandibular asymmetry associated with HFM is not progressive. In these studies, progressive canting of the maxillary plane was not routinely observed (Figs. 28-5 through 28-7).

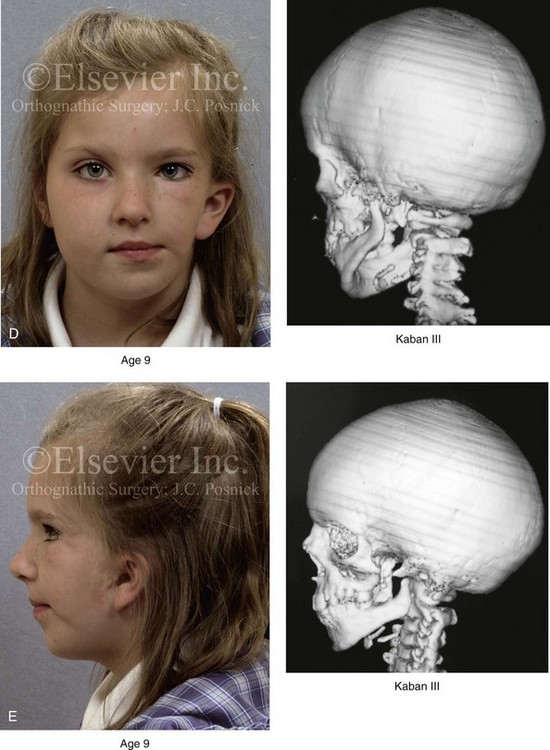
Figure 28-5 A girl who was born with left-sided hemifacial microsomia is followed up longitudinally at 5, 9, and 12 years of age without treatment intervention. There is a Type III glenoid fossa–mandibular malformation. No progression of the deformity has occurred. A, Frontal facial views at 5, 9, and 12 years of age. B, Oblique facial views at 5, 9, and 12 years of age. C, Profile views at 5, 9, and 12 years of age. D and E, Facial and computed tomography scan views at 9 years of age confirmed the extent of skeletal malformations involving the zygomatic–orbital complex, the maxilla, and the mandible.
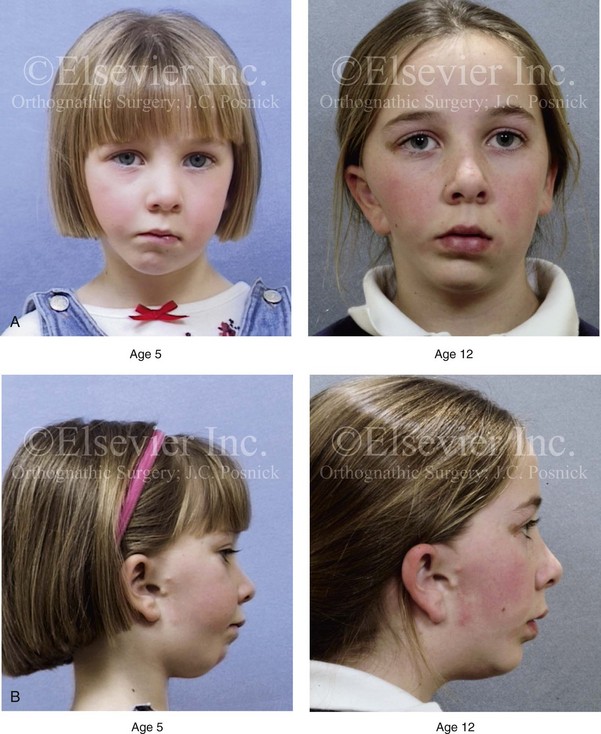
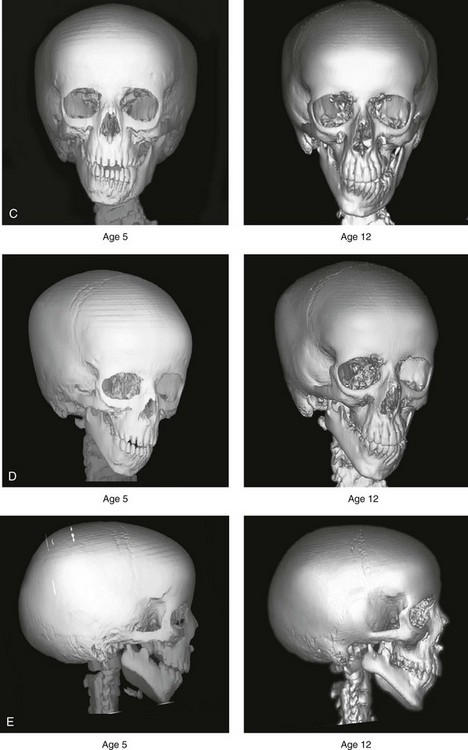
Figure 28-6 A girl who was born with right-sided hemifacial microsomia is followed up longitudinally at 5 and 12 years of age without treatment intervention. There is a Type IIB–mandibular malformation. No progression of the deformity is demonstrated. A, Frontal facial views in repose at 5 and 12 years of age. B, Profile views at 5 and 12 years of age. C, D, and E, Computed tomography scan views at 5 and 12 years of age that confirm no progression of the skeletal malformation.
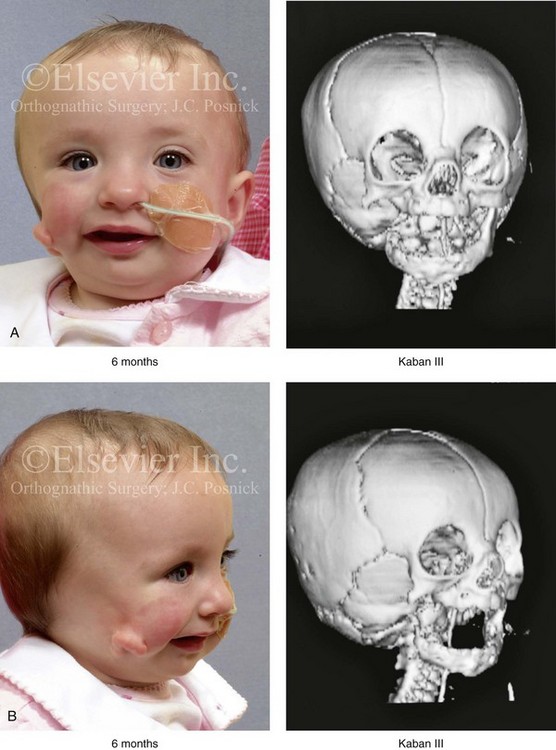
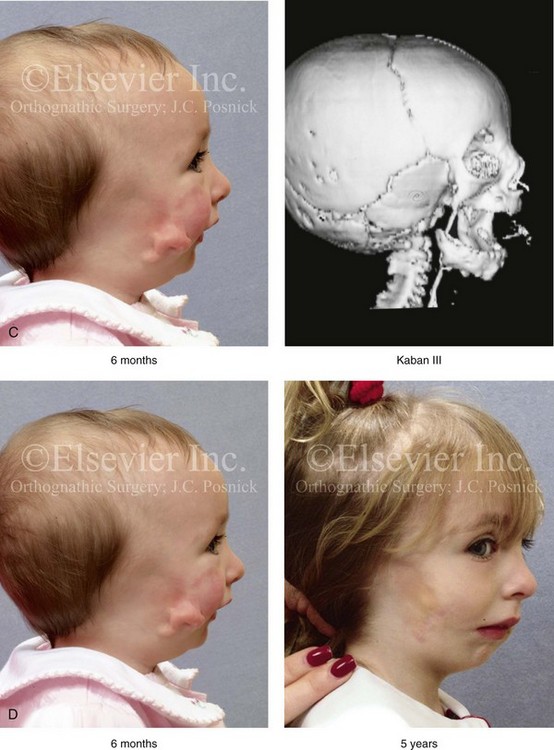
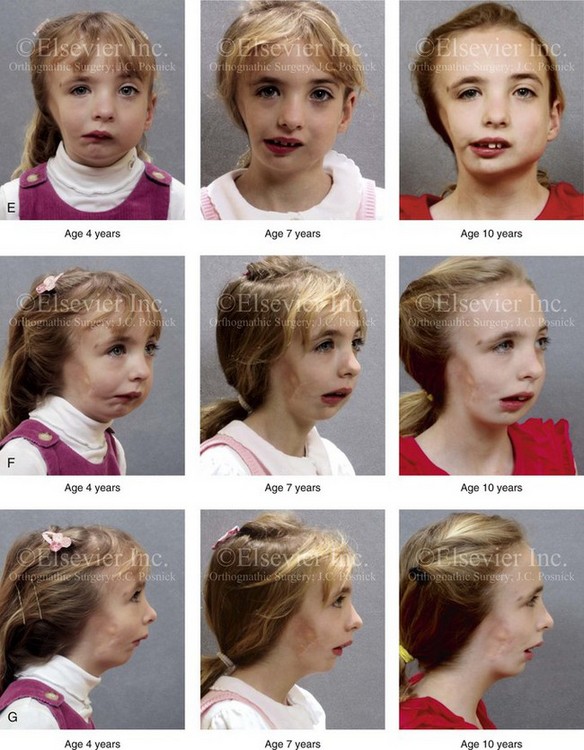
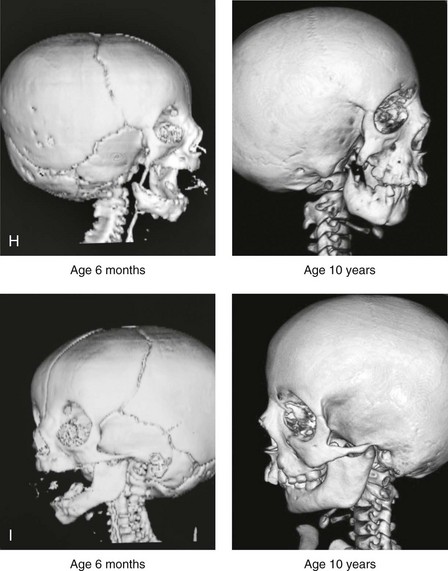
Figure 28-7 A child who was born with hemifacial microsomia of the right side. The extent of the soft-tissue and skeletal deficiencies of the structures within the first and second branchial arches is shown. There is a Type III glenoid fossa–mandibular malformation. The only treatment intervention was the excision of displaced redundant auricular tissue in the right cheek region. No progression of the skeletal malformation occurred between 6 months and 10 years of age. A, Frontal facial and computed tomography (CT) scan views at 6 months of age. B, Oblique facial and CT scan views at 6 months of age. C, Right profile and CT scan views at 6 months of age. D, Profile views at 6 months and 5 years of age. The excision of right cheek redundant soft tissue was carried out at 1 year of age with facial nerve preservation. E, Frontal views at 4 years, 7 years, and 10 years of age without treatment intervention. F, Oblique facial views at 4 years, 7 years, and 10 years of age without treatment intervention. G, Profile views at 4 years, 7 years, and 10 years of age without treatment intervention. H and I, Left and right CT scan profile views at 6 months and 10 years of age.
An alternative perspective is presented by Kaban and colleagues.92,93,95 Their research suggests to them that, as a result of ongoing growth after birth in patients with HFM, there is progressive canting measured at the pyriform rims, the occlusal plane, and the intergonial angles, especially with the Type IIB and III mandibular malformations. The authors recommend that mandibular reconstruction during the mixed dentition be considered to correct the lower jaw deformity in anticipation of normal ongoing longitudinal growth; to prevent secondary maxillary deformities (in an otherwise normal maxilla); and to alleviate psychosocial issues that would otherwise occur during childhood. I believe that Kaban and colleagues’ rationale for first-stage mandibular reconstruction during the mixed dentition in children with HFM are overly optimistic.
1. In perspective, even if a small degree of asymmetric jaw growth occurs during childhood in the patient with HFM, significant progressive dysfunction (i.e., in the areas of chewing, breathing, speech, lip control, or swallowing) that would justify a first-stage mandibular procedure is not generally seen.
2. The effectiveness of a first-stage mandibular procedure during the mixed dentition as a definitive long-term maxillofacial reconstruction has not been documented. Not only would a successful initial lengthening and derotation of the mandible be required, but the fabrication and then the periodic modification of an effective removable acrylic splint to create and maintain an ipsilateral posterior open bite (to allow for the supra-eruption of the maxillary posterior teeth) would be needed. The necessity of constant clinician monitoring and splint modification in conjunction with ongoing orthodontic treatment and rigorous patient compliance would be essential. This requires an experienced and dedicated orthodontist, a persistent surgical coordinator, and a zealous patient and family. All of the treating clinicians and the family must be in close geographic proximity and without socioeconomic barriers to treatment. Unfortunately, this is not a combination of events that is readily achievable.
3. Unfortunately, the costochondral graft construction of the neocondyle–ascending ramus required for the treatment of HFM Type IIB and III mandibular deformities carried out during the mixed dentition will result in a significant percentage of poorly growing mandibles (i.e., overgrowth or undergrowth). At minimum, a second definitive orthognathic reconstruction will be required (see the section to follow in this chapter about costochondral grafting).
4. Researchers have documented that, when a distraction osteogenesis (DO) approach is used as a first-stage mandibular procedure for HFM during the mixed dentition, symmetric lower jaw growth does not occur. The resulting residual mandibular asymmetry and deformity will, at minimum, require later definitive orthognathic reconstruction. In addition, the completion of a mandibular ramus–body osteotomy and DO during the mixed dentition is documented to result in the destruction of ipsilateral developing permanent molars in a significant percentage of children who have undergone such an operation.
 NOTE: In this author’s view, the benefits of mixed-dentition first-stage mandibular procedures rarely outweigh the downsides. The argument that first-stage mandibular surgery during the mixed dentition is necessary to allow the child to develop normal self-esteem is well intended but not realistic. Multiple doctor visits and procedures give a child a “sick kid” mentality and draw more attention to his or her differences.199 The unfortunate reality is that, for a majority of HFM patients even with well-done mixed dentition first stage mandibular reconstruction, the residual external ear, soft tissue, and other skeletal malformations will continue to draw attention to the child’s facial disproportion. The overall burden of care for the patient, the family, and the health care system is minimized when definitive single-stage maxillomandibular reconstruction is carried out at the time of early skeletal maturity (i.e., between the ages of 13 and 15) in conjunction with orthodontic treatment.
NOTE: In this author’s view, the benefits of mixed-dentition first-stage mandibular procedures rarely outweigh the downsides. The argument that first-stage mandibular surgery during the mixed dentition is necessary to allow the child to develop normal self-esteem is well intended but not realistic. Multiple doctor visits and procedures give a child a “sick kid” mentality and draw more attention to his or her differences.199 The unfortunate reality is that, for a majority of HFM patients even with well-done mixed dentition first stage mandibular reconstruction, the residual external ear, soft tissue, and other skeletal malformations will continue to draw attention to the child’s facial disproportion. The overall burden of care for the patient, the family, and the health care system is minimized when definitive single-stage maxillomandibular reconstruction is carried out at the time of early skeletal maturity (i.e., between the ages of 13 and 15) in conjunction with orthodontic treatment.
Classification of Temporomandibular Joint–Mandibular Malformation
The extent of temporomandibular joint (TMJ)–mandibular deficiency is an important factor when considering the timing of and techniques for reconstruction.108,118,119,249 Imprecise definitions of the degrees of TMJ–mandibular anomalies have resulted in miscommunication among clinicians and in the literature. In an article published during the late 1960s, Pruzansky classified the presenting mandibular anomalies of HFM into three grades (Type I through Type III) in accordance with the extent of hypoplasia of the mandibular condyle and ramus.220 Kaban and colleagues refined the Pruzansky classification to further clarify the degree of glenoid fossa–condyle–ascending ramus malformation observed in patients with HFM.94,157 The Kaban classification provides an excellent starting point for defining the reconstruction required in each individual patient (see Fig. 28-4).
A Type I mandible has only a minimal degree of hypoplasia of the glenoid fossa, the condyle, and the ascending ramus (see Fig. 28-4, A). All of the skeletal components are present, all of the masticatory muscles are present, and function is within normal limits. There is a degree of asymmetric mandibular retrognathia, with a shift of the chin off of the facial midline. A limited degree of maxillary cant can usually be documented. Note: With Type I malformations, the condyle and the glenoid fossa are fully intact. There is no need to construct a new condyle or a new glenoid fossa.
A Type IIA mandible has a moderate degree of glenoid fossa and condyle–ascending ramus hypoplasia (see Fig. 28-4, B and C). By definition, the extent of hypoplasia results in the TMJ complex being located anteriorly and medially as compared with the normal side; however, joint function remains satisfactory. The masticatory muscles have a variable degree of hypoplasia. The mandible is retrognathic, the chin is shifted to the ipsilateral side, and an anterior open bite is frequently seen. A moderate maxillary cant is usually present. With a Type IIA malformation, despite significant hypoplasia, a decision to retain the TMJ complex (i.e., the condyle and the glenoid fossa) as part of the maxillomandibular reconstruction has been made.
The Type IIB mandible involves severe hypoplasia of the condyle–ascending ramus (see Fig. 28-4, D). The glenoid fossa has an anterior and medial location, but its placement is adequate for the construction of a neocondyle. The hypoplastic condyle, if it is present at all, is rudimentary and not substantial enough to seat into the glenoid fossa. There is not a consistent “posterior stop” to the mandible against the glenoid fossa. The mandible functions with rotation but not with a condylar component in the glenoid fossa, and there is little or no translational jaw movement. Vertical mouth opening is present (albeit reduced), with a shift of the chin to the ipsilateral side. The extent of mandibular dysmorphology, asymmetric retrognathia, and anterior open bite is marked. Variable but definite deficiencies of the masticatory muscles are present. There is usually significant hypoplasia of the ipsilateral maxilla, with obvious canting. By definition, with a Type IIB mandibular deficiency and deformity, the condylar remnant is too far displaced and rudimentary to be useful. Construction of a neocondyle–ascending ramus into the current acceptable glenoid fossa is required.
The Type III mandible is free-floating on the ipsilateral side, with no “posterior stop” of the lower jaw against the skull base on the affected side (see Fig. 28-4, E). The condyle and all or part of the ramus on the ipsilateral side are absent. The disc, the TMJ capsule, and the glenoid fossa are also not developed. Mandibular dysmorphology is severe and includes ipsilateral retrognathia and decreased posterior (ramus) facial vertical height. The masticatory muscles are severely hypoplastic, and the lateral pterygoid remnant is not attached to the mandibular structures. There is significant hypoplasia of the maxilla, with obvious canting. Occasionally, a tracheostomy is required shortly after birth to manage the airway compromise that results from a combination of factors, including (but generally not limited to) the degree of mandibular hypoplasia. By definition, with a Type III glenoid fossa–mandibular malformation, the eventual construction of both a neo–glenoid fossa and a neocondyle ascending ramus will be required to achieve successful maxillofacial reconstruction.
Staging of Skeletal Reconstruction: Timing and Techniques
The facial rehabilitation of the patient with HFM must address unique and specific components of the malformation that may include the following: 1) the zygomatic and orbital region; 2) the maxillomandibular regions; 3) the facial soft tissues; 4) the external ear; 5) the auditory canal; and 6) the middle-ear structures.*
Some clinicians have speculated that there is a reduced facial growth potential in patients with HFM which then leads to a progression of the primary malformation and secondary deformities of the maxillofacial skeleton. This assumption has led some to advocate early mandibular reconstruction to prevent progression. This author agrees with Rune and colleagues,225,226 Polley and colleagues,194 Meazzini et al.136A and Ongkosuwito and colleagues161A that the observed facial asymmetry in patients with HFM is not progressive with ongoing growth (see the section earlier in this chapter about facial growth potential with HFM) (Figs. 28-5 through 28-7).203,209–212,233,234
Zygomatic and Orbital Reconstruction
The benefits of thoughtfully timed and meticulously executed orbito–zygomatic reconstruction for the patient with HFM are often underappreciated.209–213 When upper face (i.e., orbito–zygomatic) reconstruction is needed, it is carried out through a coronal scalp incision. Correction of the upper face skeletal deformity may require the intracranial disassembly and reconstruction of the dysmorphic ipsilateral anterior cranial vault; the squamous portion of the temporal bone; and the lateral orbit regions with complete construction of the zygomatic complex, including the glenoid fossa. The use of full- and split-thickness cranial bone grafts is required (Fig. 28-8).
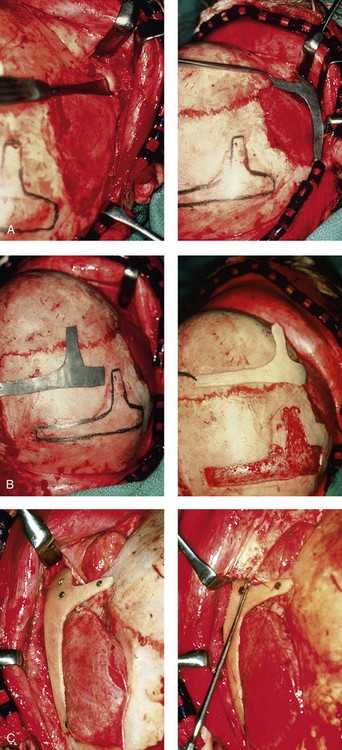
Figure 28-8 Intraoperative views of a 7-year-old child demonstrate the technique of zygomatic–orbital reconstruction as described in this chapter. When working through a coronal scalp incision, the zygomatic and orbital structures are exposed. A full-thickness cranial bone graft is harvested, and a zygomatic complex is crafted from this. The crafted zygoma is inset and stabilized with microplates and screws. The orbital defects are reconstructed as indicated with cranial bone graft, and the orbital rim is reshaped with a rotary drill. A lateral canthopexy is completed through the coronal incision and fixed to the new frontozygomatic suture region. A, View of the dissected rudimentary zygomatic remnant. A template of the proposed zygomatic complex had been made. B, The template is taken to the temporoparietal region of the skull, and its outline is marked out for craniotomy. Through a craniotomy, a full-thickness cranial bone graft is harvested for zygomatic complex reconstruction. C, The full-thickness cranial bone graft is fixed in place with microscrews. The lateral canthus is identified through the coronal incision, and a 28-gauge stainless steel wire is passed through it. The lateral canthus is pexied to the new frontozygomatic region. From Posnick JC, Goldstein JA, Waitzman AA: Surgical correction of the Treacher-Collins malar deficiency: quantitative CT scan analysis of long-term results, Plast Reconstr Surg 92:12-22, 1993.
This author believes that the reconstruction of the cranial vault, malar, and orbital deficiencies before the age of 7 years is not indicated unless functional disability warrants it.213 After the patient reaches this age, cranio–orbitozygomatic skeletal development is nearly mature. When indicated, an adult-sized anterior cranial vault, orbit, and zygomatic complex may be constructed and matched with the contralateral normal side with little concern about how future growth will alter the initial results achieved. Donor site skull reconstruction is also simpler after the age of 7 years than it is in younger children, because the presence of bicortical cranial bone will facilitate the splitting of the inner and outer tables for efficient reconstruction. Artificial bone material may also be used to reconstruct the graft donor site of the posterior skull (see Chapter 27).
Other surgeons have recommended variations in the methods and timing of the reconstruction of the upper face skeletal anomalies associated with HFM.31 Posnick and colleagues confirmed that the construction of the complete zygomatic complex with full-thickness autogenous cranial bone maintains volume and shape better than onlay grafts, which have been universally disappointing. Over time, onlay autogenous bone grafts that have been placed in the craniofacial region (e.g., supraorbital ridge, zygoma, anterior maxilla, angle of mandible, chin) from all tried donor sites (e.g., skull, hip, rib) have demonstrated significant and unpredictable resorption. Furthermore, no commensurate growth (i.e., volume expansion) of the graft with the underlying bone (e.g., the zygoma) should be expected. Some feel that, even if the onlay graft partially resorbs, at least something has been gained; however, this philosophy should be abandoned, because surface irregularities result in secondary deformities that further detract from the baseline malformation.
Maxillomandibular Reconstruction
An essential consideration when choosing the timing and techniques for maxillomandibular reconstruction in a patient with HFM is an understanding of the presenting TMJ–mandibular anatomy.2,6,7,20,25,30–32,43,46,59,96,97,180,209–212,226 The classification of the TMJ–mandibular malformation, which was described earlier in this chapter, is clinically relevant and facilitates communication.94,157 In the patient with HFM with either a Type I or Type IIA malformation, the basic maxillomandibular skeletal asymmetry and dysmorphology that requires reconstruction include the following: 1) degrees of altered facial height (i.e., decreased posterior facial height on the ipsilateral side); 2) diminished horizontal projection (i.e., deficiency more prominent on the ipsilateral side); and 3) decreased facial width (i.e. deficiency on the ipsilateral side). These malformations frequently result in canting (i.e., roll orientation) of the pyriform apertures, the maxilla, and the gonial angles; the shifting of the maxillary, mandibular, and chin midlines off of the facial midline (i.e. yaw orientation); clockwise rotation of the occlusal plane (i.e., pitch orientation); and an asymmetric class II malocclusion often with anterior open-bite malocclusion. According to the classification described, a patient who presents with a Type IIB malformation requires the construction of a neocondyle. For the individual with a Type III malformation in addition to this, there is a need for the construction of a neo–glenoid fossa.
Type I and IIA mandibular malformations are best reconstructed after all of the permanent teeth have erupted and orthodontic goals have been reached (see Fig. 28-4, A, B, and C). Surgical objectives can be met by making use of sagittal split ramus osteotomies of the mandible in combination with Le Fort I osteotomy (often in segments) and osseous genioplasty. This combination represents standard techniques, and it does not require bone grafts. The success of reconstruction is dependent on approximating mirror-image symmetry and Euclidian proportions of the skeleton through orthognathic procedures.
For Type IIB mandibular malformations, costochondral graft reconstruction of the deficient condyle ascending ramus at the time of skeletal maturity remains this surgeon’s preferred approach in most cases (see Fig. 28-4, D), despite its limitations (see the section to follow concerning condylar reconstruction with the use of costochondral graft). A sagittal split ramus osteotomy is completed on the contralateral side to derotate the distal mandible. This is combined with a Le Fort I osteotomy (often in segments) and an osseous genioplasty.
A ramus osteotomy of the contralateral side of the mandible is completed first to control repositioning of the distal mandible. The distal mandible is then secured to the maxilla via intermaxillary fixation through a custom designed acrylic splint to create the preferred reorientation of the lower jaw (see Chapter 14). It may be necessary to resect the ipsilateral coronoid process before repositioning the distal mandible. The contralateral ramus osteotomy is then rigidly fixed with bicortical screws. The ipsilateral proximal mandible is reconstructed with the autogenous costochondral graft. Harvesting the rib graft from the contralateral chest wall provides the best contour for mandibular reconstruction. The fixation of the rib graft to the native distal mandible is accomplished with a titanium miniplate and 2.0-mm or 2.3-mm screws. The fixation plate extends from the graft forward along the inferior border of the body of the mandible (fig. 28-15). It is generally necessary to recontour (i.e., with a bur on a rotary drill) the outer cortex of the distal mandible before onlaying the graft. Graft placement and fixation is often carried out through an extraoral Risdon neck incision. The avoidance of intraoral incisions on the ipsilateral mandibular ramus region during this procedure may minimize the incidence of infection. The effective seating of the neocondyle in the glenoid fossa is a critical step for successful reconstruction. In some patients, the extent of associated soft-tissue and skeletal deficiency will favor use of a vascularized fibula composite flap over the use of a costochondral graft.
The Type III glenoid fossa–mandibular malformation requires the surgical construction of the congenitally missing parts (see Fig. 28-4, E). The mandibular reconstruction is generally carried out as previously described for the Type IIB deformity. When the glenoid fossa requires construction (i.e., with a Type III malformation), so will the zygomatic complex. The glenoid fossa–zygoma and orbital reconstruction are best carried out as a separate operation when the patient is at least 7 years old (see the previous section about orbito–zygomatic reconstruction) and before mandibular reconstruction. With regard to the mandibular reconstruction, the deficiency of both the condyle-ascending ramus and the associated soft tissues may benefit from a vascularized fibula composite flap rather than a costochondral graft.
Consideration of First-Stage Mandibular Reconstruction during the Mixed Dentition
Personal Perspective Concerning First-Stage Mandibular Reconstruction during Childhood
The maxillofacial malformations in patients with HFM may be severe enough that, for airway or psychosocial reasons, the clinician feels compelled to recommend first-stage mandibular reconstruction during the mixed detention (i.e., between the ages of 7 and 11 years). Reported options for first-stage mandibular reconstruction in patients with HFM generally include 1) a ramus/body osteotomy followed by DO carried out over time for Type I and Type IIA malformations or 2) costochondral grafting with the immediate construction of a neocondyle ascending ramus for Type IIB and Type III malformations. In either case, derotation of the mandible to position the chin in the facial midline with the creation of an ipsilateral posterior open bite is usually considered the objective. The reconstructive option selected (i.e., costochondral graft versus DO) should be based on the presenting malformation and then on the following: 1) the three-dimensional morphologic results believed to be achievable; 2) the anticipated ongoing growth potential after surgery; 3) the differences in perioperative morbidity among the techniques; and 4) the overall burden of treatment to the patient, the family, the clinicians, and the health care system.209–212
Interestingly, most published reports of first-stage mandibular reconstruction in patients with HFM do not fully clarify the details of the presenting dysmorphology of the native glenoid fossa and condylar components before surgery (Figure 28-9). As stated, for DO to be effective, a functional glenoid fossa and an adequate condyle must be present (i.e., in the presence of Type I or Type IIA malformation). Unfortunately, a high incidence of bony and fibrous ankylosis has been reported in conjunction with mandibular DO in HFM.49 In addition, the consistent occurance of “undergrowth” after mandibular DO reconstruction carried out during the mixed dentition was confirmed by Meazzini and colleagues.143 Those authors conducted a comparison study of long-term follow up until the completion of facial growth of two homogenous samples of children with HFM. The experimental group was treated with mandibular DO during the deciduous or early mixed dentition in an attempt to correct the mandibular deformity. The control group was not subjected to any treatment until adulthood. The experimental group included children (n = 14) who underwent mandibular ramus osteotomies with DO (mean age, 5.9 years) with a mean follow up of 11.2 years. With the use of quantitative measurements on serial panorex radiographs, the DO group was compared with the control group (n = 8). The study results document that facial proportions in patients with HFM are maintained throughout growth when no treatment is undertaken. Unfortunately, after ramus osteotomies with DO, the mandibular disproportions returned to their original level of asymmetry during growth. The authors concluded the following: 1) HFM does not progress with regard to the degree of facial asymmetry or deformity when the patient is left to mature naturally and 2) early intervention with mandibular ramus osteotomies and DO does not effectively reduce long-term facial asymmetry in the patient with HFM.
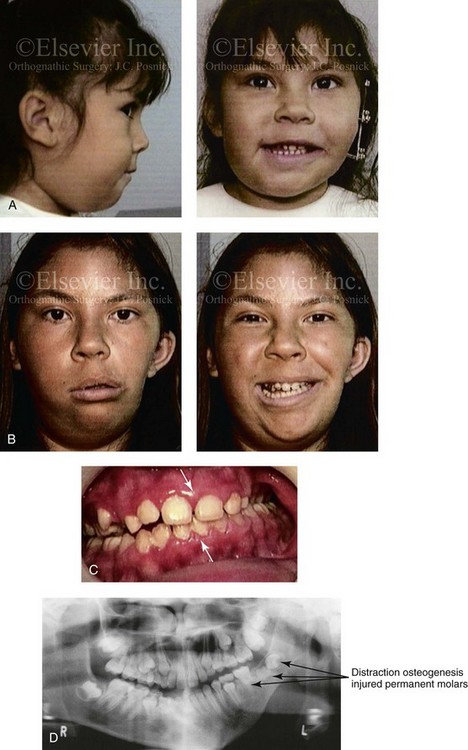
Figure 28-9 A Hispanic girl who was born with a mild form of left-sided hemifacial microsomia. The mandibular malformation was Type I. At another institution, she underwent a mandibular distraction procedure when she was 4 years old with an external device; the goals were to “derotate” the mandible, to shift the chin to the midline, and to create a posterior open bite. An acrylic splint was then used in an attempt to hypererupt the maxillary molars over time. The patient is shown at the time of procedure and then 7 years later after her referral to this surgeon. Unfortunately, the procedure carried out did not improve the occlusion or correct the facial asymmetry. During the process, the left m/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


