Treacher Collins Syndrome
Evaluation and Treatment
• Inheritance, Genetic Markers, and Testing
• Considerations during Infancy and into Early Childhood
• Dysmorphology with Treacher Collins Syndrome
• Facial Growth Potential with Treacher Collins Syndrome
• Classification of Temporomandibular Joint and Mandibular Malformation
• Staging of Skeletal Reconstruction: Timing and Techniques
• Indications for First-Stage Mandibular Reconstruction in the Newborn
• Indications for First-Stage Mandibular Reconstruction during the Mixed Dentition
• Facial Soft-Tissue Reconstruction
Treacher Collins syndrome (TCS), which is also known as mandibulofacial dysostosis, is an autosomal dominant condition with variable expressivity.* It is generally characterized by bilaterally symmetric abnormalities of the structures within the first and second branchial arches. Early descriptions are attributed to Berry,10 Treacher Collins,51 and Franceschetti and Klein.41 To date, the physical findings of many hundreds of individuals and families with TCS have been published in the world literature.
The adult patient with fully expressed TCS has a convex horizontally deficient facial profile with a prominent nasal dorsum above a retrusive lower jaw and chin (Fig. 27-1). The eye region is characterized by an antimongoloid slant of the palpebral fissure as a result of colobomata and hypoplasia of the lower lids and the lateral canthi, including the partial absence of the eyelid cilia and inferolateral orbital hypoplasia and dystopia. Tongue-shaped processes of hair frequently extend into the preauricular region. The external ears are absent, malformed, or malposed and hearing is impaired as a result of variable degrees of hypoplasia of the external auditory canals and the ossicles of the middle ears. A characteristic finding is hypoplasia of the malar bones, often with clefting through the arches and limited formation of the residual zygomas, including the glenoid fossa component. The maxilla and mandible bones are also characteristically hypoplastic, with variable effects on the temporomandibular joints (TMJs), the muscles of mastication, and the muscles of facial expression. Interestingly, hypoplasia of the centrally located soft tissues of the face is not seen with TCS. In general, there is an Angle Class II anterior open-bite malocclusion and a steep clockwise-rotated maxillomandibular complex. The posterior facial height is short, and the anterior lower facial height is long. The A-point–to–B-point relationship in profile is consistent with the clockwise rotation of the jaws. The presence of cleft palate (with or without cleft lip and choanal atresia of the nasal cavity) is variable. Dental anomalies are present in 60% of individuals; these include tooth agenesis (33.3%), enamel opacities (20%), and the ectopic eruption of the maxillary first molars (13.3%).
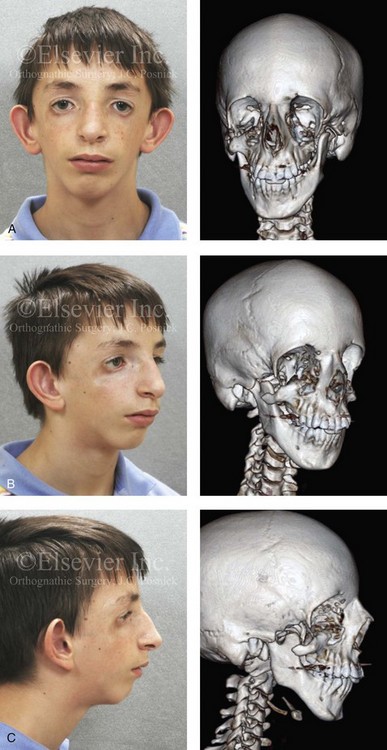
Figure 27-1 An 11-year-old boy with fully expressed Treacher Collins syndrome. His eyes are characterized by an antimongoloid slant of the palpebral fissures as a result of colobomata and hypoplasia of the lower lids and the lateral canthi, including the partial absence of the eyelid cilia and inferolateral orbital dystopia. The appearance of the malpositioned adnexal structures is a reflection of both zygomatic–orbital dystopia and hypoplasia of the soft tissues. The maxilla and the mandible are also deficient and malformed. The patient demonstrates a Type IIA mandibular malformation. The external ears are malformed and lack antihelical folds. A, Frontal facial and three-dimensional computed tomography scan views. B, Oblique facial and three-dimensional computed tomography scan views. C, Profile facial and three-dimensional computed tomography scan views.
Individuals with TCS have a reduced cranial base angle (i.e., basilar kyphosis). Craniosynostosis is not a feature of TCS, but the neurocranium may have an abnormal shape (i.e., decreased anteroposterior length and diminished bitemporal width) that is evident during childhood and remains through adulthood. 110 The degree of craniofacial malformation present at birth is believed to be relatively stable and non-progressive with age (this is discussed in more detail later in this chapter). TCS should not be confused with similar entities such as oculo–auriculo–vertebral dysplasia or Goldenhar syndrome. Distinguishing facial features of patients with TCS often include coloboma of the lower eyelids; the upper eyelids are more likely affected among patients with Goldenhar syndrome.
Inheritance, Genetic Markers, and Testing
The occurrence of TCS is in the range of 1 in 25,000 to 1 in 50,000 live births. Inheritance is autosomal dominant, and male and female individuals are equally affected.51 About 60% of probands with TCS have the disorder as the result of a de novo gene mutation. Each child of an individual with TCS has a 50% chance of inheriting the mutation (Fig. 27-2). Teber and colleagues identified TCOF1 mutations in 28 out of 36 (78%) individuals with a clinically unequivocal diagnosis of TCS.147 The most frequent clinical findings were downward-slanting palpebral fissures, hypoplasia of the zygomatic complex, hypoplasia of the mandible, conductive deafness, any degree of microtia, and atresia of the external ear canal. Although there were interfamilial and intrafamilial variations that ranged from mild to severe, there were no genotype or phenotype correlations. Four clinically unaffected parents were heterozygous for the TCOF1 mutation. The authors concluded that modifying factors are important for phenotypic expression.
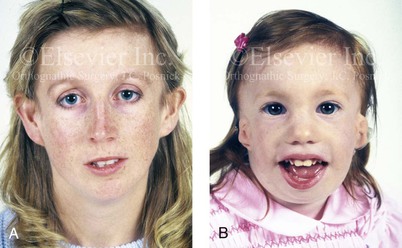
Figure 27-2 This mother and daughter demonstrate the extent of variation in the expression of Treacher Collins syndrome within a family. The mother was not aware that she carried the Treacher Collins gene until after the birth of her daughter. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997.
Genetic counseling for TCS is the process of providing individuals and families with information about the nature, inheritance, and implications of the syndrome to help them make informed medical and personal decisions. Molecular testing should be considered for any individual who presents with at least two major features or three minor features of TCS.30,32,34,62,63,72,85,88,89,154,161,165 Until recently, penetrance in TCS was believed to be complete. Marres and colleagues confirmed the absence of clinical and radiographic findings in an individual with a pathogenic TCOF1 mutation. For this reason, testing is acceptable for individuals with any degree of severity of TCS features to confirm or refute the diagnosis.
Considerations during Infancy and into Early Childhood
At the time of the birth of a child with TCS, concerns will center on the adequacy of the airway, swallowing, feeding, hearing, vision (corneal protection), the presence of cleft palate (with or without cleft lip), any other associated malformations, and the psychosocial well-being of the family.8,17,27,31,77,94,95,101,103,104,120,160 The airway may be compromised as a result of multiple factors. The first is maxillary hypoplasia, which occurs with a degree of choanal stenosis or atresia with the blockage of the nasal airway. Second, mandibular micrognathia with a retropositioned tongue that at least partially obstructs the oropharyngeal and hypopharyngeal spaces will be present. In addition, tracheomalacia, vocal cord anomalies, or neuromotor involvement may also negatively affect the upper airway. Depending on the severity of the anomalies, a spectrum of upper airway compromise may necessitate, at a minimum, special infant positioning, an extended hospital stay, and pulse oximetry monitoring. Surgical considerations for the management of airway compromise in the infant or young adult may include tongue–lip adhesion, the use of a custom-made oral appliance, immediate or delayed tracheostomy, or an urgent mandibular advancement procedure.
When a cleft of the secondary palate is documented, potential additive effects on the airway before and after repair must be considered. When Nager syndrome is present, the soft palate is both clefted and severely hypoplastic to the extent that the achievement of a functioning palate is not feasible.65,96 A primary pharyngeal flap—although helpful for velopharyngeal function—is rarely indicated, because further obstruction of the airway would likely result. Evaluation by a medical geneticist is indicated to clarify the diagnosis and all presenting anomalies, to discuss family planning issues, and to analyze chromosomal studies.
Dysmorphology with Treacher Collins Syndrome
Cranio–Orbito–Zygomatic Region
In earlier published studies, 14 reproducible cranio-orbito-zygomatic measurements taken from each of 26 standard axial CT scans of individuals with symmetric forms of TCS who had not been operated on were compared with the measurements of age-matched controls.110 The interorbital measurements (i.e., medial and lateral orbital wall separations) of the patients with TCS were at the mean when compared with their cohort group (i.e., no measurable orbital hypertelorism or hypotelorism), whereas the zygomatic measurements were significantly less than normal, thereby confirming the extent of malar hypoplasia. The congenitally deficient lateral aspect of the orbits in individuals with TCS was confirmed by the greater than normal values measured for globe protrusion and medial orbital wall protrusion in conjunction with diminished lateral orbital wall lengths. These measurements use the lateral orbital rim as a reference point and confirm the limited depth of the lateral orbits as a result of hypoplasia of the lateral orbital rims. The lateral orbital rims are components of the hypoplastic zygomatic complex. The abnormal shape of the anterior cranial vault in patients with TCS was documented as a diminished intercoronal (bitemporal) distance (width) and decreased cephalic length as compared with values for normal age-matched controls. In general, the CT measurements documented in the described study agree with the clinically observed morphology of TCS. The measurements carried out in the zygomatic structures confirmed the extent of hypoplasia of the zygomas for the group as a whole and in each individual patient; they also documented the extent of globe protrusion and decreased upper face width in patients with TCS. It appears as though the bones immediately adjacent to the involved zygomatic complex also experience a degree of hypoplasia or at least distortion.4,28,39,87,110,113,125,158,159,167,169
Maxillomandibular Region
In 1975, Roberts and colleagues completed a radiographic cephalometric study of TCS. Serial cephalometric radiographs were available for eight patients who presented with the full range of features that are characteristic of this syndrome.124 The expected facial convexity in patients with TCS was substantiated by cephalometric measurements. The extent of convexity was attributed to the severity of mandibular retrognathia (i.e., a decreased sella–nasion–B-point angle). Interestingly, the horizontal projection of the maxilla to the cranial base (i.e., the sella–nasion–A-point angle) remained within normal limits in the patients that were measured. Over time and with growth, the facial convexity remained relatively constant, thus confirming that the facial profile morphology of the infant with TCS was remarkably similar to that of the adult. Anterior facial heights were measured as “upper facial height” (i.e., the nasion to the anterior nasal spine) and “total facial height” (i.e., the nasion to the menton). The upper facial height was found to be relatively normal, whereas the total facial height was often excessive. This is the result of a combination of anterior open-bite malocclusion, mandibular retrognathism, and chin dysplasia characterized by increased vertical length and horizontal retrusion. The angle between the sella–nasion plane and the mandibular plane was obtuse, which confirmed the steepness of the clockwise rotation of the mandibular plane. The angle between the sella–nasion plane and the palatal plane was also obtuse, thus confirming the steepness of the clockwise rotation of the maxillary plane. The posterior facial height was markedly shorter for patients with TCS, with values in five of the eight patients found to be a minimum of four standard deviations less than those of the normal cohort.
The effective horizontal length of the mandible was markedly reduced, with values in most of the patients falling below four standard deviations as compared with those in the normal cohort. The decreased mandible size was evident in both the ramus height and the body length. The gonial angle was confirmed to be obtuse, with values of two standard deviations from the mean as compared with control values. The extent of antigonial notching was also documented in patients with TCS and was found to be similar to that observed among patients with condylar arrest early during their lives (e.g., as a result of juvenile rheumatoid arthritis or TMJ trauma at a young age). The steepness of the clockwise rotation of the maxillary and mandibular planes combined with the anterior and posterior vertical height disproportions and severe horizontal deficiency of the mandible is also indirectly seen in the A-point–to–B-point discrepancy. All of these morphologic findings explain the observed facial dysmorphology.4, 39,49,78,79,87
Facial Soft Tissues
Soft-tissue anomalies in individuals with TCS are manifested primarily in four clinical regions: the external ears; the eyelid–adnexal structures; the preauricular–cheek skin; and the temporal fossa (e.g., temporalis muscle hypoplasia).10,11,22,35,36,64,76,86,132,133,143,148,156 The extent of deficiency within the soft-tissue envelope is variable and, to large extent, reflective of the skeletal involvement. Each patient is unique and must be evaluated individually. The extent of soft-tissue anomalies continues to represent the most difficult obstacle to a favorable reconstruction.
External Auditory Canal, Middle and Inner Ear Structures, and Audiologic Findings
Relatively symmetric (i.e., from side to side) outer and middle ear malformations are characteristic of TCS. Middle-ear abnormalities (e.g., hypoplastic or absent cavities and ossicles) are an almost universal finding.18,55,58,66,118 The majority of patients have a unilateral or bilateral moderate or greater conductive hearing loss, with a flat or reverse sloping configuration on the audiogram. Most of the hearing loss in these patients is attributable to middle-ear malformations. In 1993, Pron and colleagues reported the results of a detailed CT radiologic and audiologic investigation of a large series of pediatric patients with TCS.118 The objectives of the study were 1) to determine the degree and symmetry of the external auditory canal and middle-ear abnormalities and evaluate hearing loss; and 2) to explore the relationship between hearing loss and external auditory canal and middle-ear abnormalities in patients with TCS. Data were available from 29 children who had not yet undergone any ear interventions and who had undergone audiometric testing and focused standardized petrous temporal bone CT scans. A review of the CT scans allowed for the evaluation of the external ear canal and the middle-ear anatomy. The majority of patients (88%) had largely symmetric external auditory canal abnormalities that were either stenotic (31%), atretic (54%), or normal (15%). Middle-ear cavity ossicular deformities were generally symmetric (96%), and cavities were either hypoplastic (85%) or missing (4%). Cavities that were hypoplastic were also deformed and assumed a rectangular rather than an oval shape. Ossicle deformities were generally completely symmetric and consisted largely of hypoplastic (46%) or missing (46%) ossicles. Ossicles (both the malleus and the incus) that were hypoplastic also tended to be ankylosed (82%) to either the lateral or medial wall of the tympanic recess. The structures of the external auditory canal and the middle ear are generally related. Increasing degrees of external ear canal malformations (i.e., normal, stenotic, or atretic) are directly associated with ossicle and cavity malformations. Of the patients with atretic canals, 67% had missing ossicles, and 33% had small or ankylosed ossicles. No abnormalities were noted in the inner-ear structures (i.e., cochlea, vestibule, canals, and internal auditory meatus) in the patients with TCS who were studied. According to the study by Pron and colleagues, hearing loss for the patient with TCS ranges from mild to severe (average, 58 dB of hearing loss).118 The majority of individuals (96%) have a moderate or greater degree of hearing loss.
Relationships exist between the external auditory canal, the middle-ear ossicles, and the degree and configuration of hearing loss.18,55,58,66,118 There is an association between the degree of external ear canal malformation and the degree of hearing loss in patients with TCS. In addition, hypoplastic, ankylosed, and missing ossicles appear to be associated with increasing levels of hearing loss; on average, the hearing loss is 52 dB, 56 dB, and 60 dB of hearing loss, respectively.
Facial Growth Potential with Treacher Collins Syndrome
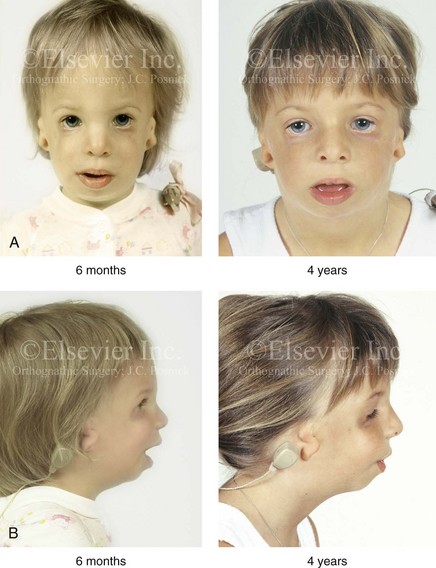
The degree of malformation present at the time of birth in a newborn with TCS is believed to be relatively stable and non-progressive with age (Figs. 27-3 through 27-6).53,102,109,114,115,124 Roberts and associates reviewed serial cephalometric radiographs from individuals who were known to have TCS.124 They documented the stability of the mandible, which continued to grow but not to catch up. Through independent research, Garner reported findings of the cephalometric analysis of three patients of varied ages who were confirmed to have TCS.49 He also documented the relative stability of the anomalies without the progression of deformity at various ages.
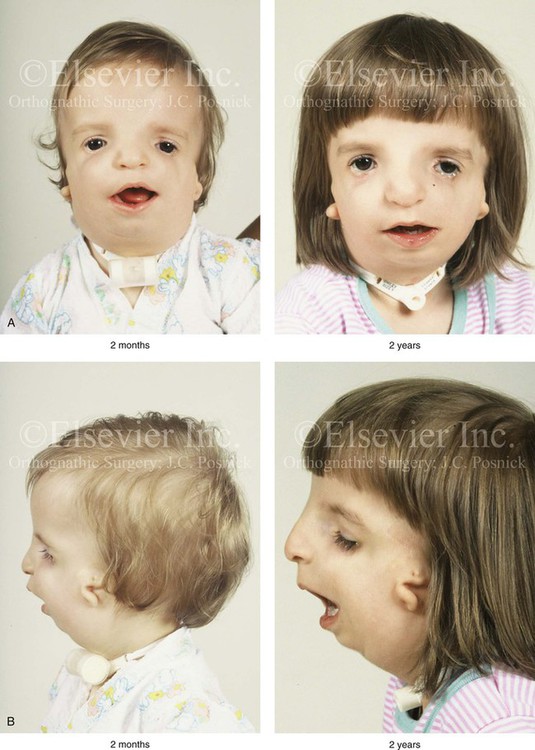
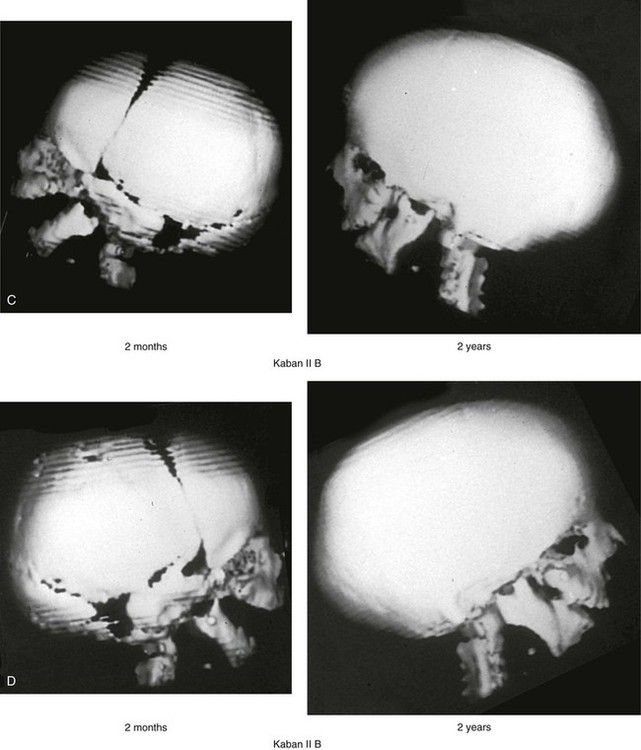
Figure 27-3 A child with a severe form of Treacher Collins syndrome was followed up longitudinally without treatment intervention. No progression of the deformity was seen during clinical or computed tomography scan examination. A, Frontal and B, profile views at 2 months and 2 years of age. C and D, Computed tomography scan views of the craniofacial region at 2 months and 2 years, respectively, demonstrating the consistent growth of the malformed condyles, the coronoid processes, and the ascending rami of the mandible. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997.
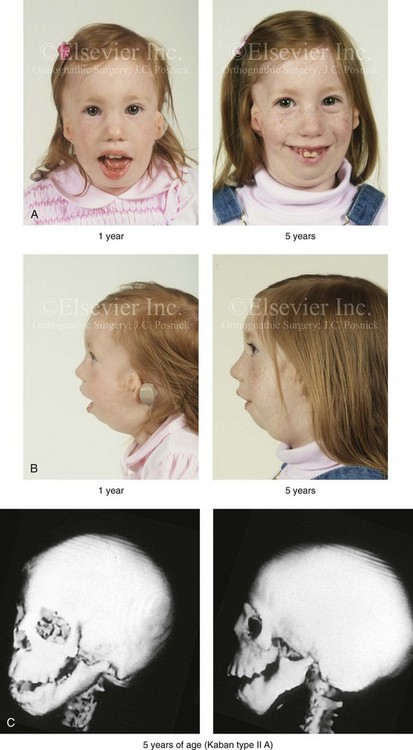
Figure 27-4 A girl who was born with Treacher Collins syndrome was followed up longitudinally at 1 and 5 years of age without treatment intervention. There is consistent growth of the craniofacial region without progression of the deformity. As a result of chronic nasal obstruction and an open-mouth posture, excess vertical facial growth can be seen. A, Frontal views at 1 and 5 years of age. B, Profile views at 1 and 5 years of age. C, Computed tomography scan views at 5 years of age. There is severe hypoplasia of the zygomatic complex with lateroinferior orbital deficiency. The mandible demonstrates a Type IIA malformation.
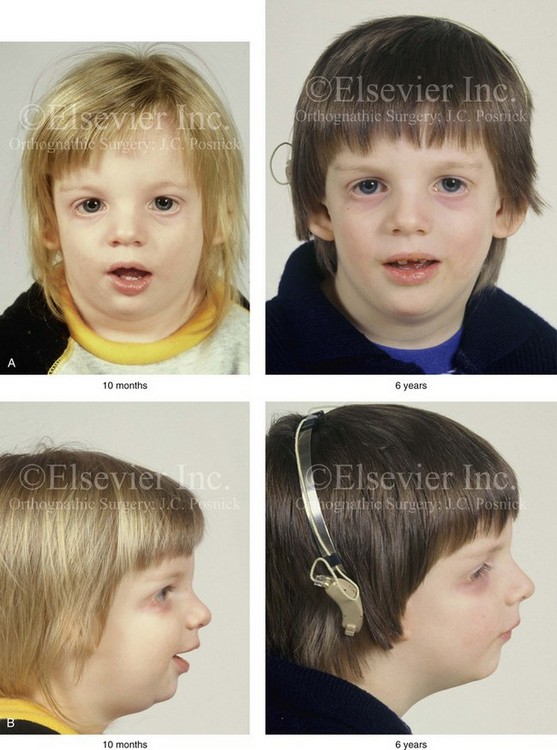
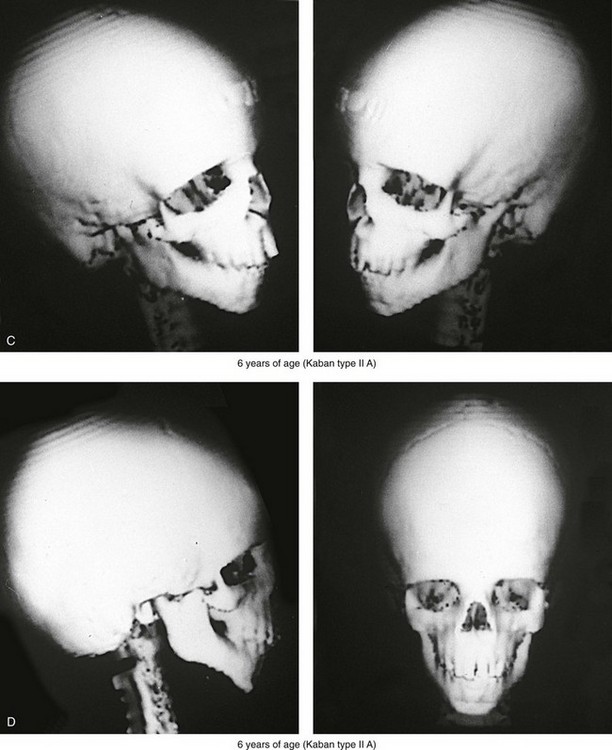
Figure 27-5 A child who was born with Treacher Collins syndrome was followed up longitudinally without treatment intervention. No progression of the facial deformity occurred with growth. A, Frontal views at 10 months and 6 years of age. B, Profile views at 10 months and 6 years of age. C, and D, Computed tomography scans.
Classification of Temporomandibular Joint and Mandibular Malformation
The extent of TMJ–mandibular malformation in patients with TCS will influence the timing and techniques of reconstruction.69,137 A lack of clinically relevant definitions of the presenting glenoid fossa–condyle–ascending ramus malformations has resulted in confusion and miscommunication. Kaban and colleagues revised and clarified the previously described Pruzansky classification system for the degree of TMJ–mandibular malformation observed in patients with hemifacial microsomia.69,119 This classification is also useful to define the presenting anomalies and then to direct reconstruction in the patient with TCS (Fig. 27-7).
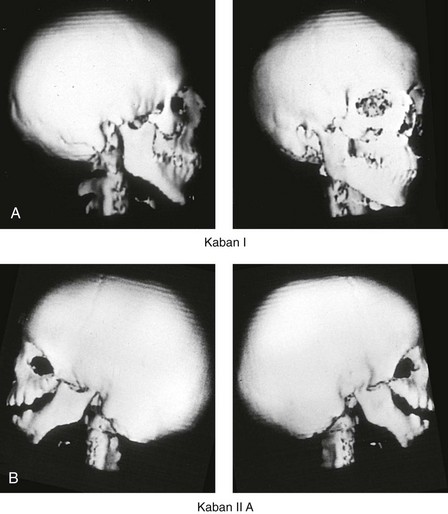
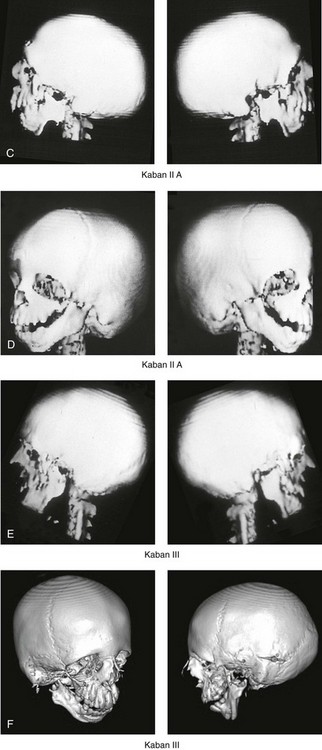
Figure 27-7 Kaban and colleagues described a classification system to define the degree of temporomandibular joint–mandibular malformation observed in patients with hemifacial microsomia. By definition, a Type IIB condylar deficiency will require the construction of a neo-condyle. The Type III malformation will also require neo-glenoid fossa construction. The same classification system is used for Treacher Collins syndrome. Craniofacial computed tomography (CT) scans demonstrate the classification system. A, CT scan views of a Type I mandibular malformation. B, C, and D, CT scan views of a Type IIA mandibular malformation. E and F, CT scan views of a Type III glenoid fossa–mandibular malformation.
A Type I TMJ–mandibular malformation involves minimal hypoplasia of the glenoid fossa and the condyle–ascending ramus on each side. All of the skeletal components are present (see Fig 27-7, A). Mandibular retrognathia and a mild anterior open bite are likely to be present, but TMJ function is essentially normal. The muscles of mastication are intact, and the condyle and the glenoid fossa are also fully intact. There is no need or advantage to constructing a neo-condyle or neo-glenoid fossa.
A Type IIA TMJ–mandible malformation involves a moderate degree of glenoid fossa and condyle–ascending ramus hypoplasia (see Fig. 27-7, B, C, and D). The anatomic location of each joint is anterior and medial, but TMJ function remains satisfactory. One or more of the paired muscles of mastication are likely to be hypoplastic. By definition, in Type IIA malformations, the condyle and the glenoid fossa have function that is at a level deemed better than could reliably be achieved through TMJ replacement. Even in patients with more severe Type IIA malformations, when applying superior and posterior pressure to the chin, the condyles can be seated into a consistent location against the skull base–glenoid fossa. By completing ramus osteotomies, the proximal segments can be positioned into a stable location against the skull base, and the distal mandible can then be reoriented into the preferred location. This will avoid the need to construct neo-condyles (e.g., costochondral grafts). Coronoidectomies may be required to accommodate the short condyles.
The Type III glenoid fossa–mandibular malformation involves deficiency to the extent that no posterior stop of the lower jaw against the cranial base is possible (see Fig. 27-7, E). The condyles and the ascending rami are not present. The disc, the TMJ capsule, and the glenoid fossa on each side are also not developed. Severe mandibular dysmorphology is present, including horizontal retrognathia and decreased posterior ramus height. A tracheostomy is generally required shortly after birth to manage the airway. Eventual construction of both a neo-glenoid fossa and a neo-condyle ascending ramus will be required on each side. Reconstruction with bone grafts (e.g., cranial grafts for the zygomatic complex and either costochondral or a vascularized fibula flap for the mandible) will be required (see discussion to follow).
Staging of Skeletal Reconstruction: Timing and Techniques
The reconstruction and rehabilitation of the Treacher Collins malformation must address unique and specific components of the anomalies, including the following: 1) the zygomatic and orbital region; 2) the maxillomandibular region; 3) the nasal region; 4) the soft-tissue envelope; 5) the external ears; 6) the external auditory canals; and 7) the middle-ear structures (Figs. 27-8 through 27-10).*
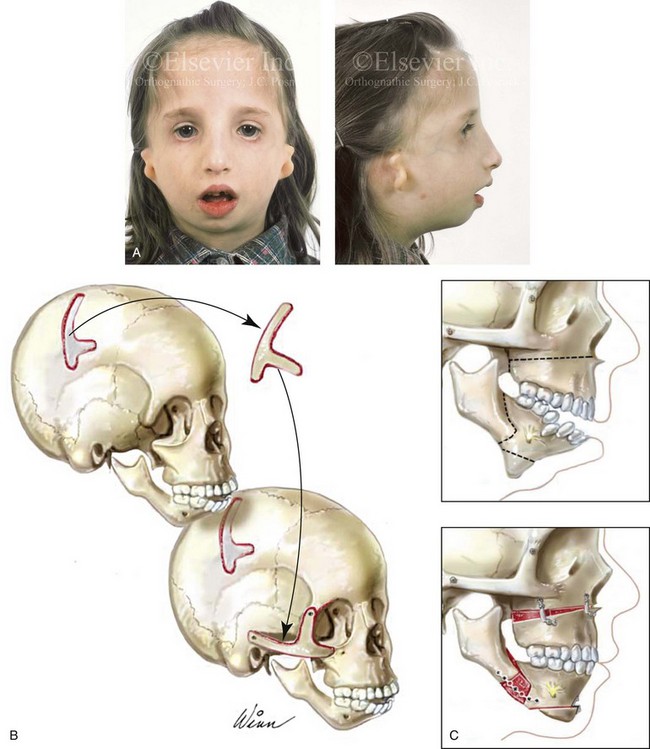
Figure 27-8 A, A child who was born with Treacher Collins syndrome is shown during the mixed dentition. She has severe zygomatic complex and orbital deficiency as well as a Type IIA condylar malformation. B, Illustration of the location of the full-thickness cranial bone graft donor site for zygomatic complex reconstruction. The zygomatic complex graft is also shown inset at the recipient site. C, Illustrations of a maxillofacial complex seen in a teenager with Treacher Collins syndrome (Type IIA mandibular malformation). The cheekbone orbital reconstruction has previously been completed. The proposed maxillary, mandibular, and chin osteotomies are marked out and then completed. Counterclockwise rotation of the maxillomandibular complex is demonstrated as part of the reconstruction.
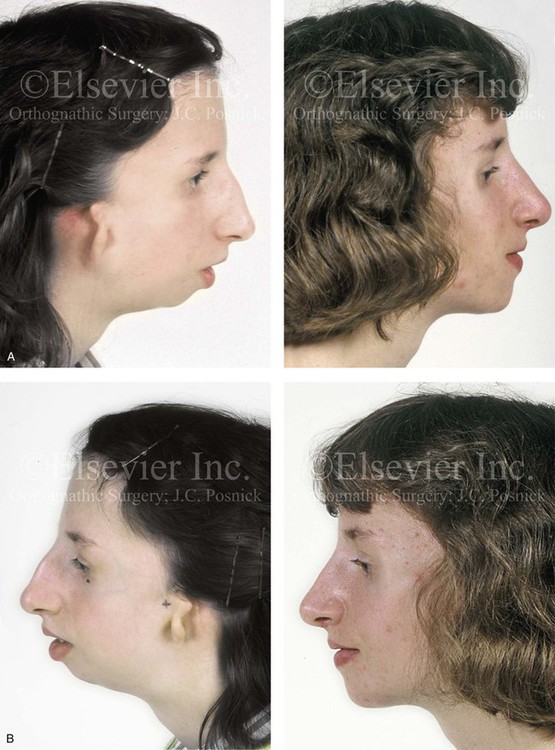
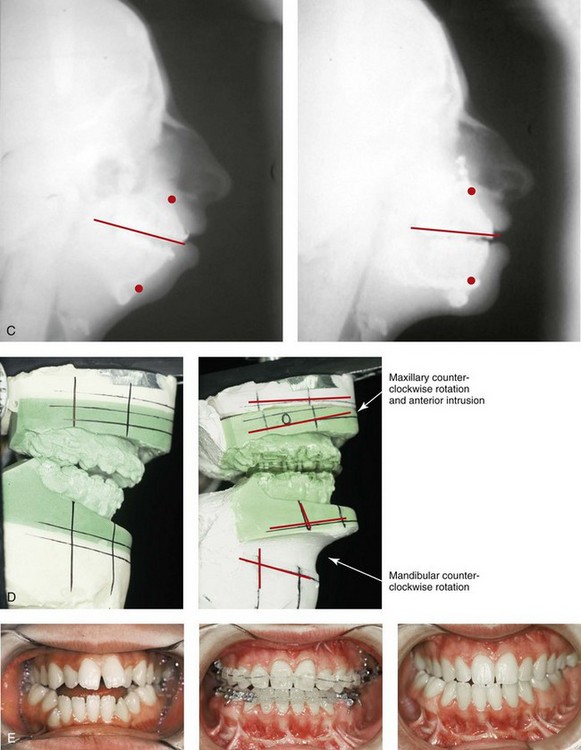
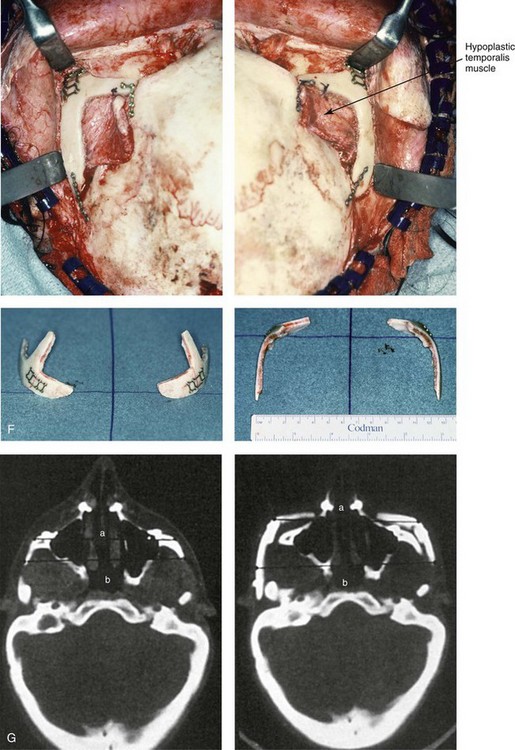
Figure 27-9 Additional images of the patient from Fig. 27-8 showing the stigmata of Treacher Collins syndrome. At the age of 17 years, the patient was referred to this surgeon and agreed to a staged reconstruction that included three operations. The initial preoperative orthodontic treatment included the extraction of mandibular first premolars with the retraction of the anterior dentition to remove compensations. Orthognathic surgery included a Le Fort I osteotomy (counterclockwise rotation and anterior maxillary intrusion); bilateral sagittal split ramus osteotomies (advancement and counterclockwise rotation); and an osseous genioplasty (vertical reduction and advancement). During a second operation, the patient underwent zygomatic complex reconstruction with fixed full-thickness autogenous cranial bone grafts as described in this chapter and as illustrated in Fig. 27-8. This was followed a third operation: septorhinoplasty (open approach) to in-fracture the nasal bones, reduce the dorsal hump (bone and cartilage), and reshape the tip, including septal (caudal strut) grafting (see Chapter 38). A, Right profile views before and after reconstruction. B, Left profile views before and after reconstruction. C, Lateral cephalometric radiographs before and after reconstruction. D, Articulated dental casts that indicate analytic model planning. E, Occlusal views before treatment, early after reconstruction, and 3 years after reconstruction. A degree of maxillary relapse (skeletal versus dental) has occurred but with a retained functional occlusion and favorable facial aesthetics. F, Crafted autogenous cranial grafts shown before and then after inset into the zygomatic defects. The advancement of the hypoplastic temporalis muscles is also demonstrated. The soft-tissue hypoplasia involves multiple layers (i.e., skin, subcutaneous tissue, fascia, and fat tissue) and therefore can not be fully corrected simply by advancing the hypoplastic temporalis muscle. G, Axial computed tomography scan views shown through the zygomas before and after malar reconstruction indicating the improved interzygomatic buttress width, interzygomatic arch width, and zygomatic arch length. A, From Posnick JC, Treacher Collins syndrome: Perspectives in evaluation and treatment, J Oral Maxillofac Surg 55:1120, 1997. F (bottom left, bottom right), G, From Posnick JC, Goldstein JA, Waitzman A: Surgical correction of Treacher Collins malar deficiency: quantitative CT scan analysis of long-term results, Plast Reconstr Surg 92:12, 1993.
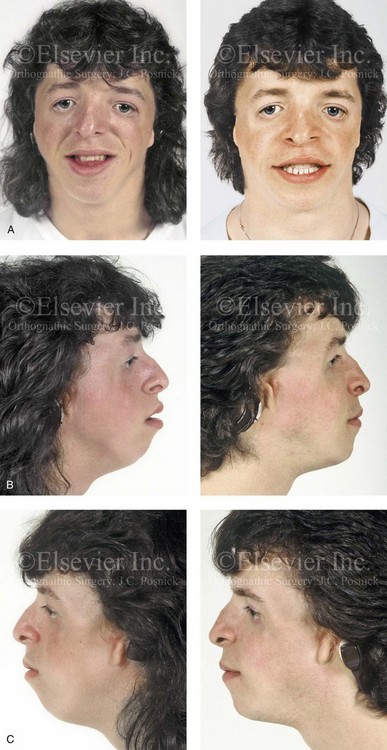
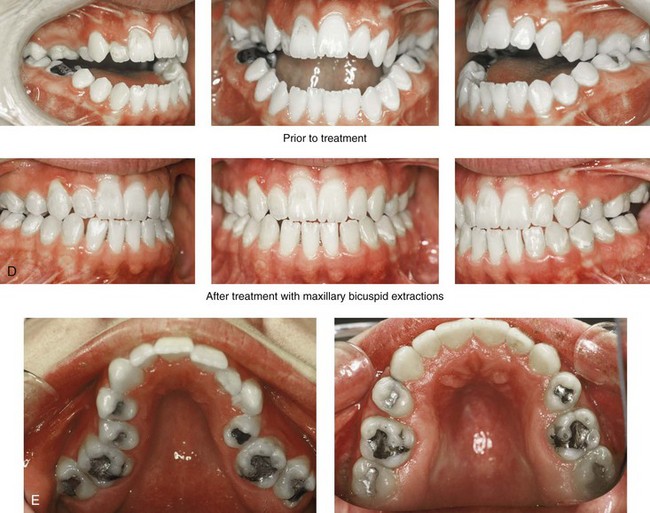
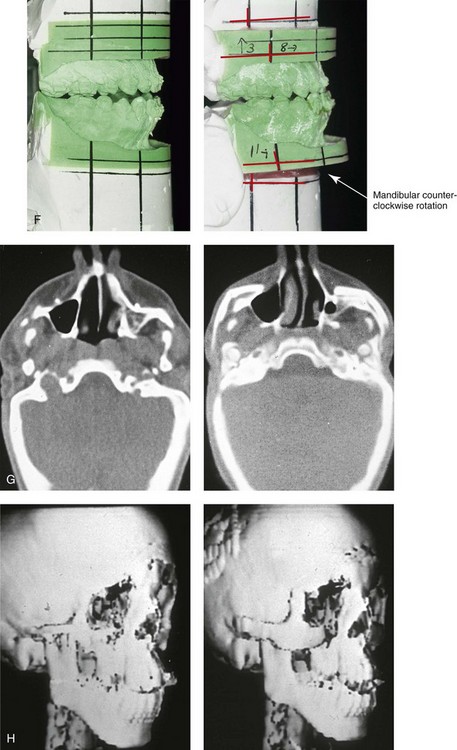
Figure 27-10 A 16-year-old boy born with Treacher Collins syndrome with a Type I mandibular malformation was followed up longitudinally to confirm no progression of the deformity. He then underwent two operations, including orbitozygomatic and orthognathic reconstruction, as described previously. In preparation for orthognathic surgery, orthodontic treatment included maxillary first bicuspid extractions with the retraction of the anterior dentition to correct the arch form. The mandibular second molars are congenitally absent. The patient’s orthognathic surgery included Le Fort I osteotomy (vertical intrusion and horizontal advancement); bilateral sagittal split ramus osteotomies (horizontal advancement); and an osseous genioplasty (horizontal advancement). He is shown before and after orbitozygomatic and orthognathic surgery. A, Frontal views before and after orthognathic surgery. B, Left profile views before and after orthognathic surgery. C, Right profile views before and after reconstruction. D, Occlusal views before and after orthognathic surgery. Note that, with the upper bicuspid extractions and the absent mandibular second molars, the maxillary second molars are no longer in occlusion. E, Palatal views before and after orthognathic surgery. F, Articulated dental casts that indicate />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


