chapter 25 Pharmacology
A variety of drugs are available for intravenous (IV) sedation. These include a number of categories, primarily sedative-hypnotics, opioids, and anticholinergics. The drugs most commonly used in IV moderate sedation are listed in Box 25-1. Also listed in Box 25-1 are several drugs (indicated by ‡) that are not recommended for IV use by dentists who have not completed a 2-year residency in anesthesiology. These drugs are discussed briefly so that the dentist may more fully understand the rationale for their not being recommended. Other drugs, such as the barbiturates, which at one time were the drugs of choice in IV sedation, have fallen from favor as newer, safer, and more effective drugs have been introduced. Discussion of the barbiturates has been minimized. For a more in-depth discussion of these drugs, the reader is directed to previous editions of this textbook.
Box 25-1
Drugs Available for Intravenous Conscious Sedation
BENZODIAZEPINES
The benzodiazepines have become the most commonly used IV sedative drugs in both dentistry and medicine. Four benzodiazepines are discussed (Table 25-1); three of them are presently available in the United States, whereas one (flunitrazepam) is available in many other countries.

Diazepam
Diazepam was synthesized in 1959 by Sternbach and Reeder. The drug became available as Valium (Hoffmann-LaRoche) in 1963 and shortly thereafter became the most prescribed oral drug in the Western world.1 Diazepam is also available in a parenteral preparation for intramuscular (IM) (see Chapter 10) and IV use. Diazepam was originally approved by the Food and Drug Administration (FDA) in November 1963.
IV administration of diazepam appears to have begun with the work of Davidau2 in Paris in 1965. This was followed shortly thereafter by a report by Main3 in 1967, who used diazepam as an adjunct to the Jorgensen technique. In 1968, Brown reported on 40 cases in which diazepam was used alone, with the drug administered until the patient felt sleepy.4
In 1969, O’Neill and Verrill5 reported on the use of IV diazepam for sedation in minor oral surgical procedures, with good to excellent results in 51 of 52 patients treated. In 1970, O’Neill et al reported on 55 patients undergoing dental surgical procedures lasting between 20 and 45 minutes. IV diazepam provided successful sedation and cooperation in 49 patients; four others moved and spoke occasionally, but were able to be treated, and two patients required additional IV medications (methohexital) for treatment to be completed successfully.6
The dosage used in these patients was that required to produce marked ptosis (drooping of the upper eyelid; Figure 25-1). Halfway ptosis of the upper eyelid is now recognized as the Verrill sign.7 The practice of administering diazepam until the appearance of the Verrill sign produces a level of sedation (central nervous system [CNS] depression) that is considered by many to be more profound than is usually necessary and is therefore not recommended for routine use.8
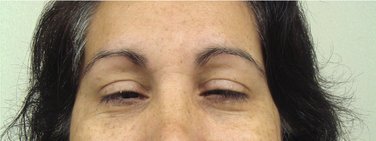
Peter Foreman,9 in New Zealand, used diazepam in combination with atropine and incremental doses of methohexital. Although successful, he stated that the addition of even small amounts of methohexital greatly increased the risk of overdose. In a subsequent study, Foreman used diazepam alone for a variety of dental therapies finding that although the degree of amnesia produced by diazepam varied significantly from patient to patient, virtually all patients agreed that dental treatment had been at least tolerable rather than an ordeal. He found that IV diazepam had made it possible to treat those patients who may not have received proper treatment in the past because of fear. Foreman stated, “Diazepam has become the drug of choice for the trained general dental practitioner, as well as for the introduction of dental students to intravenous sedation.”10
With the introduction of midazolam into clinical practice, the use of IV moderate sedation with diazepam has decreased. However, as discussed in Chapter 26, there are still occasions to consider diazepam as a first-line agent for IV moderate sedation.
Chemistry
Diazepam is a member of the 1,4-benzodiazepine group of compounds. The chemical formula for diazepam is 7-chloro-1,3-dihydro-1-methyl-5-phenyl-2H-1,4-benzodiazepin-2-one. It is a pale yellow-white crystalline powder with virtually no odor. It is considerably soluble in chloroform and acetone, moderately soluble in ethanol and ether, and poorly soluble in water.11
General Pharmacology
It is believed that emotions are largely controlled by the limbic system (i.e., the portion of the brain composed of the amygdala, hippocampus, and septal areas).12 The midbrain reticular formation, hypothalamus, and thalamus are also involved with the experience or transmission of emotions.
In very small doses, diazepam appears to act on the hippocampus, whereas other areas of the brain remain unaffected and the patient remains alert.13 Following oral administration of diazepam, this action of the drug would be appropriate; however, when diazepam is administered intravenously, a greater effect is normally desirable. Administered to the point at which sedation and ataxia (loss of muscular coordination) occur, a more generalized depression of the CNS is observed.
Research suggests that the anxiolytic properties of benzodiazepines are mediated by increased inhibitory nerve transmission.14 γ-Aminobutyric acid (GABA) is an important inhibitory neurotransmitter in the brain. Glycine (aminoacetic acid), the simplest nonessential amino acid, may be the major inhibitory transmitter of the spinal cord. The anticonvulsant and sedative properties of benzodiazepines may result from a direct agonist effect on stereospecific benzodiazepine receptors, which in turn facilitate the inhibitory action of GABA on its own postsynaptic receptors.
Fate of Intravenous Diazepam
Following IV administration, diazepam reaches a peak blood level in approximately 1 to 2 minutes. The onset of clinical activity is therefore quite rapid.15 Blood levels of approximately 1.0 µg/ml may be achieved after an IV dose of 10 to 20 mg of diazepam.16 Clinically, this would equate with a deeper level of moderate sedation (not deep sedation) and a period of anterograde amnesia.
As mentioned in Chapters 7 and 10, diazepam has a plasma half-life of approximately 30+ hours.17 A commonly held misconception is that a drug with a long half-life will possess a long duration of action, whereas one with a shorter half-life will have a shorter duration of action. This is untrue, and diazepam is an excellent example of this. The β-half-life is an indicator of the rate at which a drug undergoes biotransformation (in the liver for diazepam), whereas the factor most responsible for a drug’s duration of action is its degree of receptor-site (protein) binding.
Phase 4: 46 to 60 minutes.
At this time after receiving diazepam, virtually all patients will feel and look recovered. This is not a result of the β-half-life of the drug (30+ hours), but because of redistribution: α-half-life. The blood level of diazepam at 60 minutes after IV administration of 20 mg is 0.25 µg/ml.16 The patient is not recovered at this time. Under no circumstances should the dentist ever believe that this patient is capable of operating a car or leaving the dental or surgical office unescorted.
A clinically significant phenomenon can arise at this point, a result of the diazepam stored in the gallbladder and intestinal walls. Known as the rebound effect or second-peak effect, it involves a recurrence of symptoms of sedation and drowsiness approximately 1 hour after the first meal taken after the patient leaves the treatment site.18 In most cases, this will occur about 4 to 6 hours following the drug’s administration (start of the procedure). After a meal, particularly one rich in lipids, the gallbladder constricts, releasing its contents of bile and unmetabolized diazepam into the small intestine, where over the next hour or so diazepam is reabsorbed back into the cardiovascular system. In some patients, the diazepam blood level may reach a level at which clinical signs and symptoms of sedation recur: The patient feels quite tired and will want to lie down for a few minutes. It becomes absolutely essential therefore that the patient receiving diazepam and his or her escort be advised of this possibility before their discharge from the dental office. The rebound effect is less likely to be observed in a patient whose gallbladder has been removed.
Biotransformation
Diazepam is biotransformed by one of two pathways. In the first, the diazepam molecule undergoes demethylation to desmethyldiazepam, which possesses anxiolytic, but not sedative, effects. Desmethyldiazepam is too lipophilic to permit its excretion by the kidney. Desmethyldiazepam has a half-life of 96 hours and eventually undergoes hydroxylation to oxazepam, another pharmacologically active drug.19
Oxazepam is still another water-insoluble anxiolytic benzodiazepine. It is used as an anxiolytic agent by the oral route of administration. The pharmacology of oxazepam (Serax) is discussed in Chapter 7. The half-life of oxazepam ranges between 3 and 21 hours. It is rapidly biotransformed into its major metabolite, oxazepam glucuronide.
Effects of Age and Disease
It is often stated that drug dosages should be decreased in very young and elderly patients in addition to patients with significant liver disease. The pharmacokinetics of diazepam have been well studied in these groups of patients.20–23 The following is presented as a summary of that research:
In elderly patients, the dose of diazepam by the IV (or any other) route should be decreased for several reasons. The rate at which the diazepam undergoes biotransformation is decreased in older patients. In addition, when administered orally, the drug is absorbed in the gastrointestinal tract somewhat more slowly. However, the most important reason for the apparent increased sensitivity of older patients to diazepam (and other drugs) is related primarily to protein binding. Older patients exhibit decreased protein binding of drugs.20 This means that there will be more of the free, unbound drug available within the blood to cross the blood-brain barrier and produce CNS depression. Diazepam is offered as an example: In the younger patient, diazepam is approximately 98.5% protein bound.21 Therefore the clinical effects of diazepam are produced by only 1.5% of the dosage administered: the non–protein-bound diazepam. In the older patient in whom protein binding has decreased, diazepam may be 97% protein bound, still a significant figure, but one permitting 3% (or twice as much) non–protein-bound diazepam to be available to produce CNS depression. It becomes obvious that when administered the same dose of the drug, the clinical actions on the older patient will be exaggerated. The dosages of diazepam by the oral and IM routes must be decreased in older patients. With IV administration, titration will provide effective sedation at what will likely be a smaller dose of the drug than is usually given.
Skeletal Muscle Relaxation
Diazepam and other benzodiazepines produce skeletal muscle relaxation. Research has demonstrated that the muscle-relaxant properties of benzodiazepines are caused by central rather than peripheral effects.24 Monosynaptic reflexes, such as the knee jerk, are essentially unaffected by even large doses of diazepam, whereas polysynaptic reflexes are depressed by rather small doses.
Anticonvulsant Activity
In one study, the seizure threshold for lidocaine-induced tonic-clonic seizure activity was 8.5 mg/kg.25 When IM diazepam was administered 60 minutes before treatment in a dose of 0.25 to 0.5 mg/kg, the seizure threshold was elevated to 16.8 mg/kg of lidocaine.26
In the management of generalized tonic-clonic seizures, the benzodiazepines have not supplanted phenytoin and phenobarbital as oral maintenance anticonvulsants; however, IV midazolam or diazepam are drugs of choice in the management of status epilepticus and acute seizure activity.27 Once the seizure has been controlled, maintenance therapy with other anticonvulsants is initiated.
Cardiovascular System
Hemodynamic studies show that diazepam produces little effect on the cardiovascular system of healthy human subjects.28 IV diazepam, in a dose of 0.3 mg/kg, produces no clinically significant changes in either blood pressure or cardiac output.
Diazepam has been compared with thiopental as a preanesthetic induction agent in the cardiovascularly compromised patient (American Society of Anesthesiologists [ASA] 3 and 4).29 Administered intravenously in a dose of 0.2 mg/kg, less than 1% of the patients studied experienced a reduction of cardiac output of more than 15%, and none had a mean blood pressure reduction of more than 15%. In contrast, on receiving 2 mg/kg of thiopental, 85% of the patients exhibited more than a 15% reduction in cardiac output, whereas 68% demonstrated more than a 15% reduction in blood pressure. Adverse hemodynamic effects attributable to the benzodiazepines are rare in humans, even in patients with significant cardiac or pulmonary disease.30
Respiratory System
All sedative-hypnotics, including the benzodiazepines, are potential respiratory depressants. When these drugs are studied in patients without pulmonary disease, respiratory depression produced by intravenously administered benzodiazepines is barely detectable.31 In addition, and quite significantly, the benzodiazepines do not potentiate the respiratory-depressant actions of opioids.32
Hepatic Disease
Agitation and combativeness are occasionally encountered in patients with liver disease. Murray-Lyon et al33 in a study of patients with severe parenchymal liver disease administered diazepam intravenously.33 Adequate sedation was achieved in all patients with no deterioration of their clinical status. Diazepam, administered with care, is an appropriate sedative for patients with impaired liver function.
Pain
In general, studies have failed to demonstrate specific analgesic properties of the benzodiazepines; however, large doses of these agents will impair motor response to painful stimulation.34 These studies show that benzodiazepines are much more capable of attenuating the emotional response to pain than of altering the actual sensation of pain.
More recent studies have demonstrated that diazepam may possess some slight analgesic properties.35 These findings do not, however, alter the fact that in clinical situations in which pain control is a factor during dental treatment, local anesthetics must still be administered in the usual manner.
Amnesia
Intravenously administered diazepam produces anterograde amnesia (i.e., a lack of recall occurring from the time of injection onward).36 Retrograde amnesia, a lack of recall of events occurring before drug administration, is quite rare. Amnesia after diazepam IM administration is uncommon and is essentially nonexistent after oral administration.
Warnings
Probably the most significant side effect of intravenously administered diazepam is the occurrence of venous thrombosis, phlebitis, local irritation, or swelling. Although these complications are rare with the administration of IV diazepam as recommended in Chapter 26, a manufacturer of diazepam recommends the following as a means to minimize this possibility37:
Other warnings include the following:
The administration of IV diazepam as recommended in Chapter 26 takes into account these warnings. Titration will prevent accidental overdose in the preceding situations.
Use in Pregnancy
Any drug that crosses the blood-brain barrier also crosses the placenta, entering into the fetus. An increased risk of congenital malformation associated with the administration of benzodiazepines during the first trimester of pregnancy has been suggested in several studies.38 Because the administration of these drugs in dentistry is rarely a matter of urgency, their use at this time cannot be recommended. The possibility that a woman of childbearing potential may be pregnant at the time diazepam is used should always be considered.
Precautions
When diazepam is combined with other psychotropic agents, careful consideration must be given to possible potentiation of the drug effect.37 Categories such as the phenothiazines, opioids, barbiturates, monoamine oxidase inhibitors (MAOIs), and other antidepressants are included.
Adverse Reactions
The most frequently reported adverse reaction to intravenously administered diazepam is phlebitis at the site of injection. This is discussed in Chapter 27. Other, less frequent, adverse reactions include the following:
Paradoxical reactions such as acute hyperexcited states, anxiety, hallucinations, increased muscle spasticity, rage, and stimulation are also seen. The general term for this phenomenon is emergence delirium. It is seen more frequently with scopolamine administration and is discussed thoroughly in Chapter 27.
Dosage
The following directions regarding recommended dosage are taken from the diazepam package insert37:
Availability
Valium (Roche): 5 mg/ml in 2-ml ampules, 10-ml multiple-dose vials, and 2-ml preloaded syringe. Injectable diazepam consists of the following ingredients37:
Diazepam is classified as a Drug Enforcement Administration (DEA) Schedule IV drug. Propylene glycol and ethyl alcohol are included because diazepam is lipid soluble and relatively water insoluble; therefore it requires a nonaqueous-solvent system. Many of the complications and side effects attributed to diazepam, especially phlebitis, are in fact produced by the propylene glycol, which is closely related to ethylene glycol, a major component of antifreeze.39
Dizac (Ohmeda Pharmaceutical) is diazepam in an emulsion form that does not produce the same burning sensation as is often noted with the diazepam formulation described previously. It is available as 2.5-, 5-, and 10-mg/ml injectable solutions.40
Midazolam
Synthesized in 1975 by Walser and Fryer at Hoffmann-LaRoche, midazolam was available in many parts of the world in the early 1980s and was released for use in the United States in 1986. Midazolam first received FDA approval in December 1985. The chemical formula is 8-chloro-5(2′-fluorophenyl)-1-methyl-4H-imidazo (1,5-α)(1,4) benzodiazepine maleate. It is a colorless crystal in an aqueous solution. Each milliliter contains either 1 or 5 mg midazolam maleate buffered to a pH of 3.3.41 The acidic pH maintains the benzodiazepine ring in an open configuration, which is required for its water solubility (the diazepam ring is closed, and it is insoluble in water). Once in the body, the physiologic pH (7.4) acts to close the ring, providing the chemical structure of the drug that is required for its clinical efficacy.
Pharmacokinetics and Biotransformation
Midazolam undergoes metabolism in the liver by hydroxylation into three major metabolites.42 Whereas the major metabolites of diazepam are pharmacologically active anxiolytics, the major metabolites of midazolam have no pharmacologic activity. In addition, because of its lack of active metabolites and shorter half-life, a rebound effect is not evidenced with midazolam.
The α-half-life (distribution and redistribution) of midazolam has been recorded as 4 to 18 minutes. The β-half-life (metabolism and excretion) is 1.7 to 2.4 hours. By contrast, diazepam’s β-half-life is 31.3 hours.43 The shorter half-lives of midazolam make the drug more suitable for ambulatory sedation procedures: a relatively short duration of action combined with a relatively rapid inactivation and excretion of the drug.
Midazolam is 94% protein bound, the binding occurring primarily in serum albumin. Midazolam possesses a relatively rapid onset of action, the induction of general anesthesia having ranged from 55 to 143 seconds.44
Amnesia
Midazolam, like the other parenteral benzodiazepines, has the ability to produce anterograde amnesia. Conner et al45 demonstrated the incidence of amnesia in patients receiving IV midazolam (Table 25-2).45
Table 25-2 Incidence of Amnesia in Patients Receiving Intravenous Midazolam
| Time After Injection (min) | Amnesic Patients (%) |
|---|---|
| 2 | 96 |
| 30 | 87.5* |
| 32 | 69 |
| 43 | 57 |
* Data from Fragen RJ, Caldwell NJ: Awakening characteristics following anesthesia induction with midazolam for short surgical procedures, Arzneimittelforschung 31(12a):2261-2263, 1981.
The results shown in Table 25-2 indicate that midazolam is superior to other benzodiazepines or IV drug combinations in providing anterograde amnesia. In one study, 71% of the patients did not recall being in the recovery room.45 Other studies have not demonstrated these same remarkable results, but in all cases, the degree of anterograde amnesia provided by midazolam was at least equal to that produced by diazepam.46 Retrograde amnesia is not produced by midazolam.
A young, healthy (ASA I) woman received IV midazolam and local anesthesia for the removal of three third molars. Following the 20-minute procedure, the patient appeared alert and was quite responsive to questions. Gauze packs had been placed at the sites of extraction, and the patient had been told to bite down hard on the gauze and not to swallow. She responded verbally that she would do as directed. Within 2 minutes, the patient was complaining of a lump in her throat. Observation of the mouth indicated that all gauze packs had disappeared—the patient had swallowed them. Fortunately, they were located in the esophagus and were of no great consequence. However, when questioned, the patient had absolutely no recall either of receiving the instructions given her by the dentist or of swallowing the gauze pads.47
It becomes imperative therefore for the patient to be observed much more carefully during the in-office recovery period, that special precautions be taken to prevent such events from recurring, and that postoperative instructions (verbal and written) be given to both the patient and his or her escort. The benzodiazepine antagonist flumazenil has been shown to decrease the duration of midazolam’s amnesic period.48
Cardiorespiratory Activity
Midazolam, as is typical of benzodiazepines, has minimal effect in usual doses on the cardiovascular and respiratory dynamics of the ASA 1 or 2 patient. IV doses of 0.15 mg/kg of midazolam in healthy persons have produced statistically significant, but clinically insignificant, decreases in arterial blood pressure and increases in heart rate.49 However, other researchers noted no untoward cardiovascular response with similar doses.50 Gath et al51 recommend midazolam as an induction agent for patients with ischemic heart disease because of its rapid onset of action and minimal effects on the cardiovascular system.
Diazepam and midazolam both produce the same effects on the respiratory system. Doses of 0.3 mg/kg of diazepam and 0.15 mg/kg of midazolam produced comparable depression of respiratory response to carbon dioxide (CO2) in healthy volunteers.49 It was concluded that midazolam and diazepam injected intravenously in equipotent doses depress respiration significantly and similarly. The results of the study indicate that this is mediated by direct depression of central respiratory drive rather than caused by a simultaneous depression of the muscles of respiration, although this possibility cannot be excluded. In the doses administered, equivalent to 21 mg of diazepam and 10 mg of midazolam for the typical 70-kg adult male, such a response might be expected. Since publication of this study in 1980, it has been demonstrated that equipotent doses of midazolam are approximately one fourth of the diazepam dose.
In November 1987, Roche Laboratories, the manufacturer of midazolam, sent a warning to physicians about the use of midazolam in conscious sedation.52 It stated that the administration of midazolam had been associated with respiratory depression and respiratory arrest. Guidelines for the safe administration of this agent were offered (see Dosage and Administration). These guidelines emphasized the need for the slow titration of midazolam to all patients, especially the medically compromised.
Side Effects
The most frequently noted complaint after midazolam administration is dizziness. In the study by Conner et al,45 46% of patients mentioned experiencing dizziness. Despite this, 92% stated that they enjoyed the feeling produced by midazolam, and 100% said that they would accept the drug again if they required another operation.
Dosage and Administration
When midazolam was introduced, initial reports implied that midazolam was 1.5 times as potent as diazepam. Subsequent clinical experience with midazolam has shown it to be approximately four times as potent as diazepam. The mean effective dose for 50% of subjects (MED50) for the induction of general anesthesia is 0.2 mg/kg, although significant patient variation exists.53 Clinically adequate IV moderate sedation with midazolam should always be achieved by slow titration. In its 1987 letter, Roche recommended “an initial intravenous dose for conscious sedation as little as 1 mg, but not exceeding 2.5 mg for a normal, healthy adult.”52 Lower doses are necessary for older (over 60 years) or debilitated patients and in patients receiving concomitant opioids or other CNS depressants. The initial dose and all subsequent doses should never be given as a bolus; administer over at least 2 minutes and allow an additional 2 or more minutes to fully evaluate the sedative effect. The use of the 1 mg/ml or dilution of the 5 mg/ml formulation is recommended to facilitate slower injection.52
Availability
Lorazepam
Lorazepam is a benzodiazepine with sedative and antianxiety effects. It may be administered either intramuscularly or intravenously. Chemically, it is 7-chloro-5-(o-chlorophenyl)-1,3-dihydro-3-hydroxy-2H-1,4-benzodiazepin-2-one. Lorazepam, like diazepam, is virtually insoluble in water. Although available for IV use, lorazepam is seldom used in the outpatient ambulatory patient because of the relative inability to titrate the drug and its prolonged duration of action.54 Lorazepam was approved by the FDA in September 1977.
The introduction of flumazenil offers a means of reversing the sedative effects of lorazepam at the conclusion of the procedure. However, the clinical actions of flumazenil, especially after IV administration, are shorter than the clinical actions of lorazepam, leading to a possible recurrence of sedation after the patient is discharged from the office, a potentially dangerous situation. As discussed in the section on flumazenil, antidotal drugs and complications (see Chapter 27), consideration should be given for IM flumazenil administration whenever IV flumazenil is used.
Warnings and Precautions
Patients receiving lorazepam must be warned against operating a motor vehicle or machinery or engaging in hazardous occupations for 24 to 48 hours after its administration. Dosages of lorazepam should be decreased in patients older than 50 years to minimize the risk of oversedation.55
Adverse Reactions
The most frequently noted adverse reactions to lorazepam are caused by a direct extension of its CNS-depressant properties and include the following56:
Dosage
The following directions regarding recommended dosage are taken from the lorazepam package insert:
For the primary purpose of sedation and relief of anxiety, usual recommended initial IV dose of lorazepam is 2 mg total, or 0.044 mg/kg (0.02 mg/lb), whichever is smaller. This dose will suffice for sedating most adults and should not ordinarily be exceeded in patients older than 50 years.55
Administration
Lorazepam should be diluted immediately before IV administration with an equal volume of a compatible solution. When properly diluted, lorazepam may be administered directly into a vein or into the tubing of an existing IV infusion. The rate of injection of lorazepam should not exceed 2.0 mg/min. Lorazepam may be diluted with the following55:
Availability
Lorazepam is not highly recommended for use in outpatient sedation because of its prolonged clinical action, its extreme amnesic properties, and primarily the lack of ability of the administrator to titrate the drug to clinical effect. Lorazepam is classified as a Schedule IV drug. Lorazepam is an excellent IV sedative for nonambulatory hospitalized patients for whom close posttreatment monitoring is available for extended periods.56
Flunitrazepam
Flunitrazepam is a water-soluble benzodiazepine derivative that is chemically and pharmacologically related to diazepam and other drugs of this group. The chemical formula for flunitrazepam is 5-(o-fluorophenyl)-1,3-dihydro-1-methyl-7-nitro-2H-1,4-benzodiazepin-2-one. The sedative, antianxiety, amnesic, and muscle-relaxing properties of flunitrazepam are similar to those of diazepam except that its sedative and sleep-inducing properties are more pronounced and longer lasting than those of diazepam.57 Foreman58 reported flunitrazepam to be approximately 15 times as potent as diazepam and suggested that the drug be diluted before administration to ensure precise titration.
Flunitrazepam is available in a 1-ml ampule containing 2 mg. The manufacturer suggests diluting the drug with 1 ml of sterile water for injection before use, providing a solution of 1 mg/ml.58 Foreman, however, suggests that further dilution is warranted, recommending the dilution of 2 mg (1 ml) of flunitrazepam in 9 ml of sterile water, providing a solution of 0.2 mg/ml.58
Flunitrazepam is known on the street as “roofies.” It is sometimes snorted to offset cocaine withdrawal. It has also acquired the title “the date-rape drug” because of its ability to induce anterograde amnesia, preventing the victim from recalling specific events while under the influence of the drug.59
Side Effects and Complications
The side effects and complications associated with flunitrazepam administration are similar to those of other benzodiazepines. As with most benzodiazepines, flunitrazepam is remarkably free of respiratory- or cardiovascular-depressant effects. The most frequently reported side effects associated with flunitrazepam administration are diaphoresis, ataxia, erythema, blurred vision, hypersalivation, dry mouth, weakness, hypothermia, hypoventilation, and prolonged drowsiness.60 The dosage of flunitrazepam should be decreased in elderly and debilitated patients. The use of alcohol and driving should be prohibited for 24 hours after the administration of flunitrazepam.
Flunitrazepam Sedation in Dentistry
Foreman reported on 10 patients who received IV flunitrazepam for conscious sedation.58 The dosages ranged from 1.4 to 2 mg. Treatment conditions ranged from good to excellent in 8 of the 10 patients. No patient recalled receiving a local anesthetic during treatment (though they all did receive local anesthetics) nor did they recall any of the dental treatment. They did remember being escorted to the recovery area and driven home after discharge from the office.
BARBITURATES
The barbiturates served as an important group of sedative drugs in dentistry for almost 50 years. Niels B. Jorgensen, the father of IV sedation in dentistry, included a barbiturate in his technique of IV premedication, known worldwide as the Jorgensen technique.61
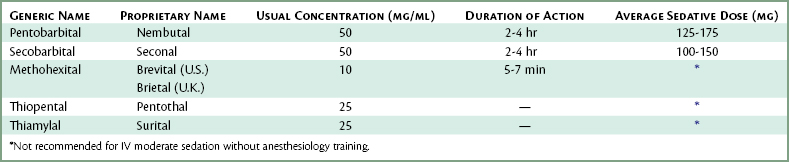
Although several barbiturates are available for IV administration (Table 25-3), only one, pentobarbital, was used to any significant degree—as a component of the Jorgensen technique.
With the introduction of the benzodiazepines, initially diazepam and now midazolam, the need for and use of barbiturates decreased to the point where, today, they are rarely indicated for use in moderate sedation. One can argue that their negative attributes (e.g., respiratory depression at therapeutic sedation levels, hangover, excitation [increased talkativeness during sedation], circulatory depression, and laryngospasm) outweighed their benefit (sedation).62
The summary section on barbiturates from the fourth edition of this text follows:
SUMMARY
Those wishing a detailed description of the barbiturates, specifically pentobarbital (Nembutal), secobarbital (Seconal), methohexital (Brevital, Brietal), sodium thiopental (Pentothal), and thiamylal (Surital) are referred to the previous edition of this textbook.63
HISTAMINE BLOCKERS (ANTIHISTAMINICS)
Promethazine, classified as a histamine blocker is, on rare occasion, employed for IV moderate sedation. The basic pharmacology of histamine-blockers has been discussed in Chapters 7 and 10. In this section, only those aspects of promethazine’s pharmacology relevant to IV administration is reviewed.
PROPOFOL
Pharmacodynamics
Central Nervous System
Propofol decreases cerebral metabolism, blood flow, and intracranial pressure.64,65 However, when larger doses are administered, marked lowering of systemic arterial pressures can significantly diminish cerebral perfusion.66
Respiratory System
Propofol, like most other IV CNS depressants, possesses respiratory-depressant properties. Propofol depresses respiration similarly to the barbiturates in normal patients (ASA 1), but to a greater degree than the benzodiazepines.67
Cardiovascular System
Propofol’s cardiovascular-depressant effects are more profound than those of thiopental.68 Both a direct myocardial depression and decreased systemic vascular resistance have been implicated in producing profound hypotension following large bolus doses of propofol.68,69 Age also affects cardiovascular response to propofol, and caution is mandatory when propofol is administered to elderly patients.70
Miscellaneous Effects
Propofol may have antiemetic effects. Studies have demonstrated an extremely low incidence of emetic sequelae after outpatient anesthesia with propofol.71 Propofol has a distribution half-life of 2 to 4 minutes and an elimination half-life of 1 to 3 hours.67
Like most IV anesthetics, propofol is eliminated via hepatic metabolism followed by renal excretion of the more water-soluble metabolites. There is some evidence that an extrahepatic route of elimination, such as the lungs, contributes to the clearance of propofol.67 Propofol is rapidly and extensively metabolized to inactive, water-soluble sulfate and glucuronic acid conjugates that are eliminated by the kidney.72 No changes in propofol’s pharmacokinetics have been reported to date in the presence of hepatic or renal disease.
Clinical Use
IV administration of propofol results in a rapid onset of action that is comparable with that of barbiturates.73,74 Recovery from propofol’s sedative-hypnotic effects is equally rapid.75 The duration of propofol’s central depressant effects increases in a dose-dependent fashion.76 In contrast to the barbiturates, there appears/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


