Chapter 22
Resin-based Filling Materials
22.1 Introduction
The development of filling materials based on synthetic polymers has been initiated by two major driving forces, in addition to the obvious commercial ones. Firstly, there was a requirement to produce a material which could overcome the major deficiencies of the silicate materials, namely, erosion, brittleness, acidity and a moisture sensitivity which demanded very careful manipulation. Secondly, developments in polymer technology produced resins which could be readily cured at mouth temperature and, with the aid of pigments and fillers, could be made to resemble the natural tooth in appearance.
The first materials to be widely used were the acrylic resins. These are, essentially, similar to resins used in denture construction (Chapter 13). The unfilled acrylic resins have now been superseded by a myriad of composite materials consisting of a heterogeneous blend of organic resin and inorganic filler.
22.2 Acrylic resins
These are supplied as a powder and liquid which are mixed together. The composition is similar to that given in Table 13.1 for denture base materials. Briefly, the materials consist of a powder and liquid. The powder contains beads of polymethylmethacrylate (<50 μm), chemical initiator (often a peroxide) and pigment, whilst the liquid consists of methylmethacrylate monomer and a chemical activator (often a tertiary amine). The pigments used are, generally, white, yellow or brown, in order to match natural tooth shades, as opposed to the pink pigments used in denture base polymers. The setting reaction involves a free radical addition polymerisation as described in Chapter 12.
The amine which is often used as a chemical activator in acrylic resins remains largely unconsumed at the end of the setting reaction. One such amine which has been widely used is N, N′ dimethyl-p-toluidine. This has very poor colour stability, particularly in ultraviolet light, and gradually changes from clear to brown, causing a consequent change of colour in the material itself; hence some acrylic filling materials have been reported as having very poor colour stability. Attempts to overcome this problem have involved the use of UV absorbers and more stable amines as well as other types of activators and initiators. Peroxide/alkylborane systems have been used as an alternative to peroxide/amine. This system was claimed to produce a material with adhesive properties although this has never been extensively evaluated.
Other materials utilized a peroxide/mercaptan initiation system whilst peroxide/sulphinic acid was used in other products. The latter system is complicated because the sulphinic acid (normally p-toluene sulphinic acid) is unstable and can inadvertently cause premature polymerisation of the monomer if incorporated in the liquid. This problem is overcome by incorporation of a salt of the sulphinic acid in the powder with the peroxide. A small quantity of methacrylic acid is incorporated in the liquid. This rapidly converts and sulphinic acid salt to the free acid when powder and liquid are mixed together. The sulphinic acid then reacts with the peroxide to activate the polymerisation process.
Advantages: The acrylic resin materials are less prone to erosion than silicates. They have low solubility over a wide range of pH values. They are less acidic than silicates though cannot be considered biologically bland due to the presence of residual methylmethacrylate monomer. They are less brittle than silicates although their mechanical properties are far from ideal.
The materials are good thermal insulators having a low value of thermal diffusivity, 1.0 × 10−3 cm2 s−1 as opposed to 2.0 × 10−3 cm2 s−1 for dentine. The ability to match the appearance of tooth substance is, initially, very good, although some products have a tendency to discolour gradually with time. Discoloration at the margins is also observed with many restorations.
Disadvantages: Although the acrylic materials do not contain any strong acids, some products contain methacrylic acid, used to modify setting characteristics, and all contain a certain level of residual methylmethacrylate monomer which is irritant. This, coupled with a significant temperature rise during setting caused by a highly exothermic polymerisation reaction, necessitates the use of a protective cavity base material. The material of choice is a setting calcium hydroxide type. Products containing eugenol should be avoided since they retard the setting of the resin and cause discoloration.
The materials undergo a considerable setting contraction (6% by volume). If uncontrolled, this could produce a significant marginal gap down which fluids could penetrate. The problem may be partially alleviated by filling the cavity with small increments and allowing the contraction to occur towards the walls of the cavity before the next increment is added. Another approach is to overfill the cavity and place the setting material under strong finger pressure with a matrix strip during setting.
The coefficient of thermal expansion value for acrylic resin is some ten times greater than that for tooth substance (see Table 22.2). The potential for percolation of fluids down the restoration–tooth interface when the patient takes hot or cold food and drink is, therefore, significant.
The mechanical properties of acrylic resin compare unfavourably with those of the natural tooth material which it replaces (Table 22.1). The low value of modulus of elasticity indicates that acrylic resin is a far more flexible material than either enamel or dentine. Flexing of restorations under load can lead to marginal breakdown. The lower compressive strength and hardness values of acrylic resin are reflected in a poor durability, particularly when restorations are subjected to abrasive forces. Material loss by wear is a phenomenon associated with these relatively soft materials.
Current status: Acrylic resins are now rarely used as permanent filling materials. They overcome the major problems associated with silicates but have many other disadvantages which preclude regular use. The materials are still in use for temporary crown and bridge construction.
22.3 Composite materials – introduction
A composite material is a product which consists of at least two distinct phases normally formed by blending together components having different structures and properties. The purpose of this is to produce a material having properties which could not be achieved from any of the individual components alone. The two main components of composite filling materials are the resin phase and the reinforcing filler. The beneficial properties contributed by the resin are the ability to be moulded at ambient temperatures coupled with setting by polymerisation achieved in a conveniently short time. The disadvantages of using resin alone have been given in the previous section. The beneficial properties contributed by the filler are rigidity, hardness, strength and a lower value for the coefficient of thermal expansion. In addition, if the filler occupies a significant proportion of the volume of a composite material it markedly lowers setting contraction. The effect of filler depends on the type, shape, size and amount of filler incorporated and, often, the existence of efficient coupling between the filler and resin.
Table 22.1 Some mechanical properties of acrylic resin, enamel and dentine.
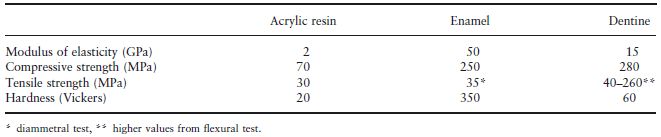
Fig. 22.1 Variation of surface hardness, setting contraction and coefficient of thermal expansion with inorganic filler content for acrylic resins.
When particulate glass fillers are incorporated in acrylic resins, three properties, namely, coefficient of thermal expansion, setting contraction and surface hardness, depend almost linearly on the filler content, as shown in Fig. 22.1.
Other thermal and mechanical properties may vary in a similar way. Strength and modulus of elasticity generally increase with addition of filler, as does abrasion resistance, probably as a result of increased surface hardness. If the added filler is translucent, the optical properties of the resin are improved and a more lifelike appearance produced.
The resins used in composite materials are invariably based in methacrylate monomers. Simple methylmethacrylate was used in some early products but most materials now utilize dimethacrylates. These monomers undergo a smaller contraction on setting and form a highly cross-linked three dimensional network with properties which are generally better than those of polymethylmelthacrylate.
Fig. 22.2 Polished surface of a dental composite showing filler particles embedded in a matrix of resin (×1445).

22.4 Classification and composition of composites
All dental composites consist of a blend of resin and inorganic filler. Methods used to characterise materials are based upon the method used to activate polymerisation of the resin and on the particle size distribution of filler. Figure 22.2 shows the polished surface of a composite in which the filler particles embedded in a matrix of resin are clearly visible.
Resins: The nature of the resin may alter slightly from one product to another, although, essentially, they all contain a modified methacrylate or acrylate. Figure 22.3 gives the molecular structures of two of the most commonly used monomers, Bis GMA and urethane dimethacrylate, together with that of tri-ethylene glycol dimethacrylate (TEGMA) which is a comonomer often used to control the viscosity of the unmixed materials. It can be seen from Fig. 22.3 that the monomer and comonomer molecules are difunctional methacrylates. Each carbon–carbon double bond is able to take part in a free radical addition polymerisation, to give a highly cross-linked resin after setting. New composites, only just appearing on the market, are based upon a different kind of chemistry in which polymerisation occurs through a ring-opening mechanism. These products are considered in the context of the reduction in setting contraction (see page 207).
Fig. 22.3 Molecular structures of three modified methacrylate or acrylate resin monomers used in composite materials. (a) Bis GMA (addition product of BisPhenol A and glycidylmethacrylate). (b) Urethane dimethacrylate. (c) Triethylene glycol dimethacrylate.
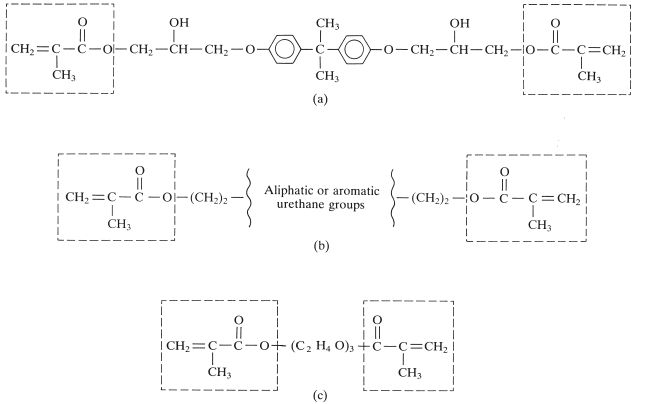
Bis GMA is a difunctional methacrylate which is normally formed by a reaction between bisphenol A and glycidylmethacrylate. It has two phenyl groups which lend a degree of rigidity to the molecule and the hydroxyl groups are thought to give some intermolecular hydrogen bonding. These features give Bis GMA a consistency which is similar to that of thick treacle at room temperature. Blending of filler particles with a material of this consistency is difficult and the manufacturers normally have to use a fluid diluent monomer such as TEGMA to reduce the viscosity. A mixture of three parts Bis GMA and one part TEGMA is typically blended with filler. Bis MA monomer is used in some products. This is very similar to Bis GMA but contains no hydroxyl groups.
The urethanedimethacrylate monomers may be aliphatic or aromatic in nature. The central group in Fig. 22.3 is typically:
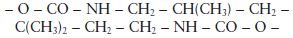
in a commonly used aliphatic dimethacrylate monomer. Monomers of this type have relatively low viscosity and do not require the use of a diluent monomer although some manufacturers do blend various monomers together. Urethanedimethacrylate monomers containing aromatic groups have a slightly more complicated structure and are often more viscous, normally requiring the presence of a diluent monomer.
The size of the monomer and comonomer molecules, coupled with a rapid increase in viscosity during setting, causes a relatively high concentration of acrylate or methacrylate groups to remain unreacted after setting. This results from the fact that reactive methacrylate groups find it increasingly difficult to migrate to the reaction sites as viscosity increases. When the glass transition temperature of the polymerizing mass approaches the ambient temperature the rate of diffusion diminishes markedly and little further reaction is possible. Further reaction could be encouraged by heating the material to a temperature well above the ambient temperature in order to regenerate mobility of reactive groups. Heat treatment of this type is not feasible for direct composite filling materials but may be used to enhance the properties of composite inlay materials.
Methods of activation: Polymerisation may be activated chemically, by mixing two components, one of which typically contains an initiator and the other an activator, or by an external ultraviolet or visible light source. The traditional method for delivering the blue visible light required for ‘visible light activation’ involves the use of a quartz tungsten halogen (QTH) lamp. Other systems including plasma arc, laser and light emitting diode (LED) systems are now also available. These various types of lamps are discussed on p. 204.
For chemical activation, many different methods of dispensation are available. The most popular is the ‘two paste’ system (see Fig. 22.4). Each paste contains a blend of resin and filler. One paste contains about 1% of a peroxide initiator, such as benzoyl peroxide, whilst the other paste contains about 0.5% of a tertiary amine activator, such as N, N′ dimethyl-p-toluidine on p-tolyl diethanolamine. The ensuing reaction is a free radical addition polymerisation as described in Chapter 12.
Other systems which rely on chemical activation are as follows:
Fig. 22.4 A typical two paste composite material. Approximately equal amounts of two pastes are taken out of the containers using a plastic spatula and then they are mixed together on a paper pad.
Light-activated materials are generally supplied as a single paste which contains monomers, comonomers, filler and an initiator which is unstable in the presence of either ultraviolet (UV) or high-intensity visible light. For UV-activated materials, the most commonly used initiator is benzoin methyl ether. At certain selected wavelengths within the UV range, this molecule is able to absorb radiation and undergo heterolytic decomposition to form free radicals. The radicals initiate polymerisation which then continues in much the same way as that described for vinyl monomers (p. 102).
The use of UV-activated materials has diminished greatly since the possible dangers of long-term exposure to ultraviolet radiation were highlighted.
For visible light-activated materials the initiator system comprises a mixture of a diketone and an amine. Camphorquinone is a commonly used diketone which rapidly forms free radicals in the presence of an amine and radiation of the correct wavelength and intensity.
Light-activated materials require the use of a specialist light source, capable of delivering radiation with the appropriate characteristics to the surface of the freshly placed material in situ. (see Fig. 22.5)
Care must be taken in the storage of unused pastes since exposure to sunlight, or particularly to the surgery operating light, may be sufficient to activate a slow initiation process which causes the paste to thicken and become unworkable. For materials which are supplied in pots it is essential to replace the lid quickly after removing the required amount of paste.
Many materials are supplied in syringes which will enable the operator or his assistant to expel sufficient material for one restoration. The material remaining in the syringe is not exposed. Another method of dispensation is in the form of ‘compules’. Each compule contains sufficient paste for at least one restoration and is extruded into the cavity by placing the compule into a press. (see Fig. 22.6)
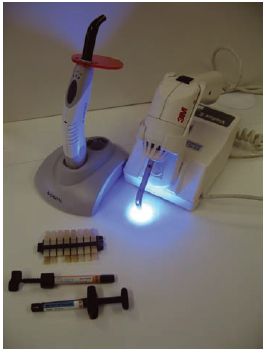
Fig. 22.5 Light-activated composite materials are often provided in light-proof syringes of the type shown here. Materials are provided in various tooth shades corresponding to the different shades on the shade guide which is also shown in this figure. The same manufacturer is likely to also supply a light curing unit which is used to activate polymerisation. The curing units shown here are a QTH unit (on the right) and an LED unit (on the left).
Fillers: The type, concentration, particle size and particle size distribution of the filler used in a composite material are major factors controlling properties.
Fillers commonly used include quartz, fused silica and many types of glass including aluminosilicates and borosilicates, some containing barium oxide.
The first generation of composite materials typically contain 60–80%, by weight, of quartz or glass in the particle size range of 1–50 μm. The particle size distribution may vary, within this range, from one product to another, some containing relatively greater amounts of larger particles, approaching 50 μm, others containing larger quantities of smaller particles. Materials containing filler particles of this type are normally referred to as conventional composites. The filler particles are subjected to a special pretreatment prior to blending with the resin. This involves laying down a surface coating of a coupling agent on the particles to enhance bonding between the filler and resin matrix. The coupling agent most commonly used is γ-methacryloxypropyltrimethoxysilane:
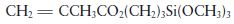
This is a difunctional molecule which at one end has the characteristics of a methacrylate monomer whilst at the other it has a silane group capable of interacting and bonding with glass or quartz surfaces. Hence, it is able to set up a bond between the resin and filler components in a composite system.
Fig. 22.6 This shows the three most common means of supplying composite filling materials. On the left we have the two paste chemically activated materials supplied in pots. Mixing of the pastes is required to bring about activation of polymerisation. In the middle we see the syringe format and on the right we see mini-syringe or ‘compule’ format. Both the syringe and compule format are used for light-activated materials which require no mixing. In the case of the compule format each small mini-syringe contains sufficient material for one or two restorations and the compule is disposed of after use.

Since the composite filling materials were introduced in the mid-1960s, there has been a trend towards the use of fillers with smaller particle size. Many of the currently available products with glass or quartz fillers contain particles in the more limited range of 1–5 μm.
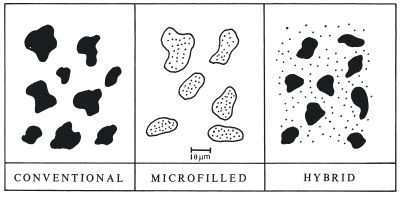
Another development has been the introduction of a number of products containing submicron particles of silica. These materials are referred to as microfilled composites and contain silica particles in the range 0.01–0.1 μm with a typical mean diameter of 0.04 μm. The very small particle size produces a massive increase in available surface area for a given volume of filler (typically 103–104 times more surface area). Consequently, it is not possible to incorporate very high filler loadings of this small particle size and products which are available contain only 30–60% filler by weight. Even at these lower levels, calculations show that many filler particles must be present as agglomerates and not as individual particles surrounded by resin.
The method of incorporating the smaller particles varies, direct blending with resin being difficult. The most widely used method is to prepare prepolymerised blocks of resin containing a high filler loading of silica. The block is splintered and ground to give particles of resin up to 100 μm in diameter, each containing silica. These particles are blended with monomer, comonomers, initiators or activators to form pastes.
A third series of composite materials contain a blend of both conventional glass or quartz particles together with some submicron, particulate silica. These products are referred to as hybrid composites. Using filler loadings of about 75% conventional size (1–50 μm) and 8% submicron size (0.04 μm average), a total filler content of 83% or greater can be achieved. Some hybrid products contain a blend of at least three different particles sizes of filler. These allow efficient packing of filler into the smallest possible volume and enable filler loadings of up to 90% by weight to be achieved.
Figure 22.7 shows diagrammatically the microstructure of the main groups of materials.
Modern formulations of composites often involve new methods of manufacture of fillers in which the particle size and shape is controlled and in which the traditional boundaries between conventional, microfilled and hybrid products can no longer be clearly identified. Wet techniques including sol-gel technology as well as jet blasting techniques in which streams of powder are fired at other streams to reduce and control particle size and shape give the manufacturer the opportunity to optimize all aspects of filler composition and nature. Composites produced using very small particles of less that one μm average diameter are often termed ‘nanocomposites’ by manufactures. However, the essential characteristics of such materials are adequately defined within the groups of microfilled and hybrid materials without the need to consider a whole new class of products.
Conventional, microfilled and hybrid composites are all available as either chemically activated or light-activated products.
The ISO Standard for resin-based restorative materials (ISO 4049) classifies materials as being of two types depending upon the intended application. Type 1 is claimed by the manufacturer to be suitable for the restoration of cavities involving occlusal surfaces. Type 2 includes all other polymer-based filling and restorative materials. The standard further sub-divides materials into three classes as follows:
- Class 1 comprises self-curing materials whose setting is activated by mixing an initiator and an activator.
- Class 2 materials are those whose setting is effected by application of energy from an external source such as blue light or heat. These materials are further sub-divided as follows:
- Class 3 materials are dual-cure materials which have a self-curing chemical mechanism but are also cured by the application of external energy.
Fig. 22.7 Diagrammatic representation of the size of filler particles used in the three main groups of composite resins. A further group of ‘nanocomposites’ have particles similar to those used in microfilled products.
Another method for classifying composite materials which has been developed and used by the manufacturers recognizes the fact that many dentists choose materials based upon their handling characteristics. Hence, some highly viscous materials are classified as ‘packable’ composites whilst some more fluid products are classified as ‘flowable’ composites. There is an understanding that the choice of packable or flowable material may vary depending upon the particular clinical application. Figure 22.8 shows a packable material which has a viscosity which is so high that it cannot be extruded through a syringe or compule and an alternative means of providing the material in small portions of paste sufficient for one restoration has been devised.
22.5 Properties of composites
Some of the properties of resin-based restoratives are included within the tests and requirements of the ISO Standard (ISO 4049). This standard sets minimum standards of quality in relation to the following:
Fig. 22.8 Some very viscous light-activated materials cannot be extruded from syringes or compules and they may be provided in containers of the sort shown here. The viscous material does not require any mixing and can be packed directly into a prepared cavity. Materials of this sort are sometimes referred to as packable composites.

Biocompatibility: Whilst composites are considered to be generally acceptable in terms of biocompatibility they should be treated as potentially harmful materials and handled with caution. The potential risks with composites are discussed in comparison with mercury in dental amalgam in Section 21.4. As with most materials, the products are potentially more harmful before setting when smaller molecules are not strongly bound within the mass of material. After setting the rigid cross-linked structure helps to bind potentially harmful components more tightly.
Setting characteristics: For chemically activated materials, setting commences immediately after mixing the two components (paste and paste or paste and liquid etc.). The rate of set is uniform throughout the bulk of the material causing a gradual increase in viscosity at room temperature. Hence, the materials have a limited working time and must be inserted into the prepared cavity before they become unmanageable. In ISO 4049 the working time is determined using a thermocouple located at the base of a small cavity (6 mm deep by 4 mm diameter). The working time is taken as the time when the exothermic heat of reaction for the mixed material causes a noticeable rise in temperature. The standard requires that the working time (timed from start of mixing) should be at least 90 seconds.
After insertion, the materials are held under pressure with a plastic matrix strip and setting is normally completed within two or three minutes. Since setting occurs uniformly throughout the material it is safe to assume that a hard surface indicates that the material has set right through to the base of the cavity. Any material which is not covered by the matrix during setting is likely to have a tacky surface layer due to inhibition of the polymerisation reaction by oxygen. In ISO 4049 the setting time is determined using the same apparatus as that described for measuring working time. For setting time determinations the equipment is maintained at mouth temperature (37°C). The setting time is defined as the time to reach the peak temperature in the exothermic setting reaction. The setting time measured by this method must be no more than 5 minutes.
For light-activated materials, only a minimal increase in viscosity takes place before the material is exposed to the activating light source. With these products the operator has, therefore, a longer working time. It should be remembered, however, that visible light-activated materials do begin to set slowly after exposure to light, particularly light of high intensity such as the surgery operating light. Therefore, insertion of the material into the cavity should not be delayed longer than necessary. In ISO 4049 light-activated materials are required to be tested for sensitivity to ambient light. When subjected to lighting equivalent to a dental operating light they should show no detectable change in consistency after 60 seconds exposure. After being covered with a matrix strip and exposed to the light source, polymerisation is often very rapid. Exposure times of between 10 seconds and one minute are, typically, required to cause setting. This ability to set rapidly after exposure to a light source is termed command setting.
The pattern of setting for light-activated materials is dictated by the fact that activation is first achieved in the surface layers of material where the light intensity is greatest. The potential for activation declines exponentially as a function of the distance from the surface of the filling. The intensity of light Ix at a distance x from the surface is given by the function
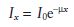
where I0 is the light intensity at the surface and μ is the absorption coefficient of the material. Since a certain level of intensity is required to cause activation it follows that light-activated materials have a limited depth of cure. The high viscosity of the pastes retards the diffusion of active free radicals from the surface layers to the lower unactivated layers, hence, material which is not activated initially may take a considerable time to set or may remain unset indefinitely. In ISO 4049 depth of cure is evaluated simply using a scrape test. Material is packed into a cylindrical mould and cured from one end for the time specified by the manufacturer. Immediately after curing, the mould is opened and any soft uncured material is removed using a plastic spatula. The height of the remaining hard, cured material is used to indicate depth of cure. In recognition of the fact that the method is quite crude and that material near the centre of the mass of material cures to a greater depth than the edges, the measured value is divided by 2 to give the final calculated depth of cure. This should be no less than 2 mm for most materials. A lower value of 1 mm is allowed for opaqueing materials.
Manufacturers of light-activated composites can control depth of cure by formulating the products in such a way that they more readily allow light penetration. In addition, they can supply or recommend a light source of adequate intensity and stipulate the exposure time required to give a certain depth of cure. Darker and more opaque shades of materials cannot be cured to the same depth as light and translucent shades. As an example, the light paste of one material can be cured to a depth of 2.5 mm using a 30 second exposure to light. The brown, opaque paste of the same material can only be cured to a depth of 1 mm using the same exposure time. Increasing the exposure time has very little effect on the depth of cure. If a material cures to a depth of 2.5 mm following a 30 second exposure it will not be cured to a significantly greater depth by increasing the exposure time to 1 or 2 minutes. On the other hand, the depth of cure can be significantly reduced by using a light exposure time of less than that recommended by the manufacturer.
The compatibility of light sources and composite materials has been the subject of several studies and debates. Most currently available light-activated composite materials utilize a similar catalyst system and most light-activation units are designed to deliver radiation which has a high intensity at the relevant wavelength. There are marked differences in performance between the units however, with a variation in intensity of light at 470 nm of up to ten times (130–1300 lux at 470 nm). Since depth of cure values which are supplied by manufacturers have normally been measured with a specific light source, it cannot be guaranteed that the same depth of cure could be achieved with a different light source.
Other factors can be controlled by the operator. The distance of the light source from the surface of the material is important. Depth of cure decreases significantly as this distance increases. The operator should never attempt to cure a greater depth of material than that recommended by the manufacturer, nor should he attempt to use a shorter exposure time. When large cavities are being restored the composite material should be cured in increments to ensure proper curing.
The polymerisation reaction which causes setting of composite materials is exothermic in nature. The heat liberated can cause a significant temperature rise within the material. If the temperature rise is excessive and the pulp is not effectively insulated, irritation, sensitivity or more serious irreversible damage may be caused. The heat of reaction for chemically activated and light-activated materials is of similar magnitude but the resulting temperature rise varies considerably. In the case of a typical chemically activated material the temperature rise is expected to be in the range 1–5°C for an average size of restoration. For light-activated materials the temperature rise is typically 5–15°C depending upon the monomer system used and the filler content. The temperature rise is much higher for light-activated materials because the heat of polymerisation is liberated over a much shorter time-scale. In addition, the heating effect of the light-activation unit further increases the temperature of the composite material when it is illuminated. In order to minimize the latter effect, manufacturers incorporate filters into their light-activation units. These filters are designed to remove the ‘hotter’ parts of the white light which occur at the red end of the visible spectrum. Hence, the radiation used in most units appears blue.
Light activation units: The purpose of the light activation unit is to deliver high intensity radiation of the correct wavelength to the surface of the material in order to activate polymerisation. The critical wavelength used by the vast majority of units and materials is 470 nm which corresponds to the blue region of the visible spectrum.
Several types and designs of light activation unit are available for activating the polymerisation of light-activated materials. The conventional type of blue visible light activation unit used in dentistry since the early 1970s (shown in Fig. 22.5) is based upon light produced by a quartz tungsten halogen bulb (often shortened to QTH or halogen bulb) equivalent to that used in motor car headlights or slide projectors.
The design of the light curing unit is either a light box with a flexible fibre-optic umbilical and a light wand that is used to illuminate the restoration or a gun design where the bulb and cooling fan are located in a hand-held device with a rigid fibre optic light guide protruding out of the front of the gun to conduct light to the restoration. There are problems with all fibre-optic systems in that the glass fibres break with time reducing the efficiency of light transmission. This is a greater problem with flexible bundles than with rigid light guides. Both designs will include a cooling mechanism for the bulb, a timing unit and filters to remove some of the unwanted spectra produced by incandescent light sources, particularly the ultra-violet end of the spectrum.
Halogen lamps are the most widely used and are available in both designs. They are capable of generating the required power of light output and are relatively cheap. The power output from the bulb deteriorates as the bulb ages reducing the effectiveness of the curing process. These bulbs produce a broad spectrum of light and require appropriate filtration to eliminate harmful elements of the light spectrum. They also generate a considerable amount of heat both directly and through emissions in the infra red range of the light spectrum.
Alternative light delivery systems: The light energy can now be delivered through other alternative means using plasma arc, laser or light emitting diode (LED) systems (Fig. 22.5). These delivery systems vary in the spectral distribution of radiation produced and in the power or intensity of radiation over the critical wavelength region. In the case of camphoroquinone the optimal absorption of radiation occurs at 460–480 nm and systems which produce high intensity radiation outside the effective region are likely to be inefficient. They may require filters to remove unwanted radiation and cooling systems to reduce the effects of some of the radiation which causes heating but is ineffective in activating polymerisation. Such is the case with QTH type lamps described above.
The plasma arc lamps use a Xenon bulb which works on the same principle as a motor vehicle spark plug. A spark is produced by generating a large potential difference across a gas which becomes ionized to form the ‘plasma’. Plasma arc lights are more commonly available in the light box/umbilical/light wand design. The power output from the bulbs is considerable (most are of the order of 300 watts) and require very efficient cooling systems. There are a smaller number of recently designed products that use a new design of light and reflector working at 120 watts that can be mounted in a gun design. The power output from these light units is about ten-fold that of a halogen bulb resulting in considerably shorter curing times and possibly greater depth of cure. There is also a significant heating effect with these bulbs through the infrared spectrum.
The total amount of light generated for plasma lamps is significantly greater than in the QTH lamps. Filtering is required to remove radiation with <400 nm or >500 nm wavelength and exposure times much shorter than those with QTH lamps are recommended. Typically, only 2 or 3 seconds of exposure is required with plasma arc lamps to achieve the same depth of cure obtained with a 30-second exposure to a typical QTH lamp. The timer on the control unit for these plasma arc lamps normally allows exposure times of only a few seconds followed by a latent period during which further exposure is not allowed. This prevents undue tissue heating which may result from over-exposure. Nevertheless, h/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


