11 Oral Manifestations of Autoimmune, Immune-Mediated, and Allergic Disorders
11.0 INTRODUCTION
An intact immune system is essential for health, and in the context of oral diseases, is critical for the prevention and clearance of infections. Many disorders are characterized by an exaggerated response of the immune system, producing inflammatory or autoimmune reactions that lead to further disease. These conditions can be broadly classified as autoimmune, immune-mediated, and allergic disorders, each being potentially associated with oral manifestations. Management of these disorders depends largely on the clinical diagnosis and severity of features, and frequently requires topical and/or systemic immunomodulatory therapies for management. Oral health providers should be aware of the potential oral manifestations of these disorders, as they can be associated with significant morbidity and in some cases may be the initial presenting feature of the disease. This chapter will describe the clinical diagnostic features of the most commonly encountered autoimmune, immune-mediated, and allergic disorders with oral manifestations.
11.1 Autoimmune conditions
Autoimmune diseases result from loss of self-tolerance or an overactive immune response and are characterized by the production of circulating autoantibodies and immune complexes. In addition to autoantibody production by plasma cells, T-cells also play an integral and critical role in the pathogenesis of autoimmune conditions. Approximately 5–8% of the U.S. population has been diagnosed with autoimmune disease, and women are more likely than men to be affected. Diagnosis can be challenging due to a myriad of signs and symptoms that can develop over months to years prior to full disease presentation. The etiology of autoimmune disorders is poorly understood, though it is likely associated with genetic, infectious, and/or environmental factors. Some conditions primarily target epithelium, resulting in vesiculobullous disorders, while others primarily affect connective tissue and are known as collagen vascular diseases. The diagnosis and evaluation of Sjögren syndrome, an autoimmune disease with significant oral considerations, is covered in detail in chapter 8.
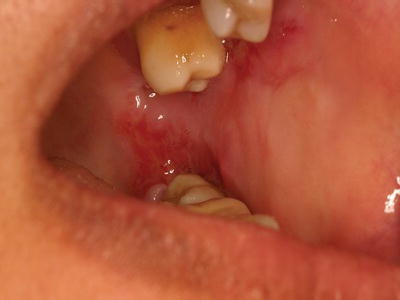
Figure 11.1 Collapsed bulla in a 52-year-old female with pemphigus vulgaris.
11.1.1 Autoimmune conditions that target epithelium
11.1.1.1 Pemphigus vulgaris
Pemphigus vulgaris (PV) is a rare autoimmune vesiculobullous condition that causes mucosal and cutaneous lesions, with oral involvement often being the first sign of the disease. If left untreated, PV can be life-threatening due to extensive skin ulcerations and infection. The skin lesions appear as blistering eruptions (vesicles or bullae) with a raw erythematous base. Any mucosal and/or skin surface can be involved, including oral as well as conjunctival, pharyngeal, and laryngeal mucosa. Oral vesicles and bullae are rarely observed as they quickly break down, resulting in large, irregular ulcerations (Figure 11.1). During clinical examination of patients with autoimmune vesiculobullous disorders, a blister can be induced secondary to soft tissue manipulation, which is referred to as a positive Nikolsky sign.
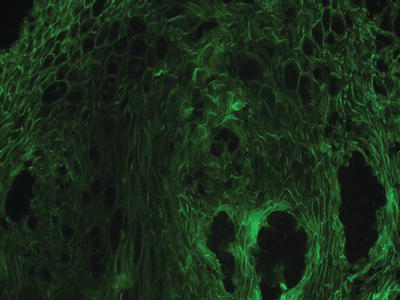
Pemphigus is characterized by the production of autoantibodies to desmogleins (integral proteins in the desmosomes that confer epithelial cell to cell adhesion), which can be detected in affected tissues (via direct immunofluorescence [DIF]; see chapter 1) as well as in the systemic circulation (via serum-based indirect immunofluorescence [IIF] or enzyme-linked immunosorbent assay [ELISA]). Intact biopsy specimens of pemphigus may reveal intraepithelial separation with preservation of the epithelial basal cell layer, often described as a “row of tombstones.” Diagnosis should be confirmed by DIF, which reveals autoantibodies (usually IgG or IgM and complement) in the intercellular spaces between the epithelial cells, resulting in the classic “fishnet” appearance (Figure 11.2). IIF evaluates circulating blood for the presence of specific immunoreactants directed against epithelial structures and may be useful in distinguishing pemphigus from pemphigoid and other oral diseases. In PV, IIF antibody titers correlate to the level of clinical disease and can be useful in assessing response to therapy. Further, autoantibody tests for levels of PV antigens desmogleins 1 and 3 are commercially available and correlate to disease activity in a similar manner as IIF findings.
Paraneoplasic pemphigus is a PV-like disease that occurs infrequently in patients with an underlying neoplasm and/or lymphoproliferative disorder, with the most frequent being non-Hodgkin lymphoma, Castleman disease, chronic lymphocytic leukemia, thymoma, sarcoma, and lung carcinoma. This disorder tends to have a severe and progressive phenotype, with extensive and painful ulcerations primarily affecting the lips, tongue, soft palate, and conjunctiva. Certain drugs like captopril and penicillamine can trigger a PV-like drug hypersensitivity reaction that reverses once the drug is withdrawn.
Figure 11.2 Direct immunofluorescence pattern of a tissue specimen from a patient with pemphigus vulgaris.
11.1.1.2 Pemphigoid
Mucous membrane pemphigoid (MMP), also known as cicatricial pemphigoid, is a vesiculobullous autoimmune disease that affects oral and other mucosal surfaces including conjunctival, nasal, and vaginal mucosa. Skin lesions associated with pemphigoid are characterized by bullae that eventually rupture, followed by crusting and healing without scarring. A significant complication of MMP with ocular involvement is conjunctival scarring (symblepharon) and blindness; therefore all patients diagnosed with pemphigoid require monitoring by an ophthalmologist. Pemphigoid is characterized by autoantibodies directed against hemidesmosomes, which connect the basal epithelium to the basement membrane and the underlying connective tissue, resulting in subepithelial separation and bulla formation. Intact intraoral blisters are observed more frequently compared with PV due to the greater thickness of the blister “roof” (Figure 11.3). Eventually bullae rupture, producing ulcerated lesions that appear clinically similar to those of PV and generally heal without scarring. MMP often presents as a desquamative gingivitis, in which the gingival epithelium spontaneously sloughs or can be easily removed with minor manipulation, resulting in erythematous and/or ulcerated gingiva.
Biopsy specimens of pemphigoid demonstrate a split between the epithelium and the underlying connective tissue, with complete separation of the basal epithelium from the basement membrane. DIF shows a continuous linear band of immunoreactants (usually IgG and C3) directed at the basement membrane zone (Figure 11.4). IIF for pemphigoid is of limited diagnostic value since circulating antibodies in serum are not consistently present and titers do not correlate with disease activity.
11.1.2 Autoimmune conditions that target connective tissue
11.1.2.1 Lupus erythematosus
Lupus erythematosus (LE) is the most common autoimmune connective tissue disorder and has a wide spectrum of disease presentation mediated by the deposition of immune complexes into various organs. Systemic lupus erythematosus (SLE) is a multisystem disease characterized by a cutaneous rash of the malar region secondary to sun exposure (with a “butterfly” appearance; Figure 11.5), renal dysfunction, musculoskeletal manifestations (arthritis and myalgias), cardiac complications (vasculitis, pericarditis, or non-bacterial endocarditis with vegetations affecting the heart valves, termed Libman-Sacks endocarditis), and thrombocytopenia (Table 11.1). Oral involvement develops in up to 25% of patients and may present as areas of lichenoid inflammation (section 11.2.2) or non-specific ulcerations that clinically resemble other common oral ulcerative conditions (i.e., recurrent aphthous ulcerations; Table 11.2).
Figure 11.3 Intact bulla on the left lateral tongue of 67-year-old female with mucous membrane pemphigoid complaining of persistent oral soreness.
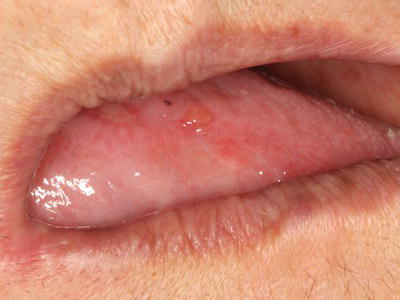
Figure 11.4 Direct immunofluorescence pattern of a tissue specimen from a patient with mucous membrane pemphigoid.
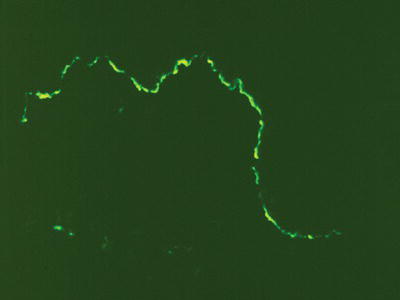
Figure 11.5 Malar rash on the cheeks of a 53-year-old female with systemic lupus erythematosus.
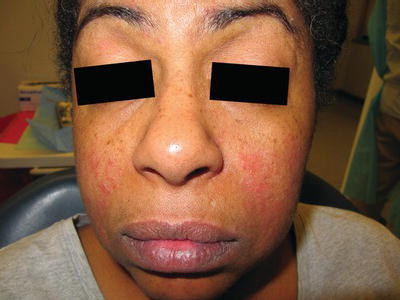
Table 11.1 American College of Rheumatology criteria for classification of systemic lupus erythematosus.*
| Presence of 4 or more symptoms simultaneously or serially on two separate occasions | |
| Sign/Symptom | Definition |
| Malar rash | Rash on cheeks |
| Discoid rash | Red, scaly patches on skin that cause scarring |
| Photosensitivity | Exposure to ultraviolet light causes rash or other symptoms of SLE flare-ups |
| Oral or nasal ulcers | Presence of oral or nasopharyngeal ulcerations |
| Arthritis | Non-erosive arthritis of two or more peripheral joints, with tenderness, swelling, or effusion |
| Serositis | Pleurisy (inflammation of the membrane around the lungs) or pericarditis (inflammation of the membrane around the heart) |
| Serology | ≥ 1 of the following in the absence of offending drug: – Hemolytic anemia – Leukopenia (WBC < 4,000 cells/μL) – Lymphopenia (< 1,500 cells/μL) – Thrombocytopenia (< 100,000 cells/μL) |
| Antinuclear antibody test | Positivity (titer ≥ 1:160) |
| Immunologic disorder | Positive anti-Smith, anti-ds DNA, antiphospholipid antibody, or positive test result for lupus anticoagulant test using a standard method; or false-positive serologic test for syphilis |
| Renal disorder | Proteinuria or cellular cases seen in urine microscopically |
| Neurologic disorder | Seizures or psychosis in the absence of offending drugs |
*Modified from http://www.rheumatology.org/practice/clinical/classification/SLE/1997_update_of_the_1982_acr_revised_criteria_for_classification_of_sle.pdf, accessed February 28, 2012.
Chronic cutaneous lupus erythematosus (CCLE) or discoid lupus is a variant of LE in which lesions are generally limited to skin and mucosal surfaces with few or no systemic signs. Skin lesions are characterized by scaly, erythematous patches that typically present on sun-exposed skin surfaces, and healing may result in scarring and hypo- or hyperpigmentation. Oral manifestations of CCLE include classic lichenoid lesions, with characteristic erosions surrounded by white, radiating striae (Figure 11.6).
Table 11.2 Clinical and laboratory findings of autoimmune, immune-mediated, and allergic conditions with oral manifestations.
| Disease | Clinical Presentation | Laboratory Findings |
| Autoimmune Disease | ||
| Pemphigus vulgaris | Intraepithelial bullae Denuded mucosae/ulcers |
LM: intraepithelial separation DIF: antibodies in intercellular spaces (fishnet appearance) IIF: circulating antibodies, correlates with disease activity |
| Mucous membrane pemphigoid | Subepithelial bullae Desquamative gingivitis Oral ulcers Symblepharon (cicatricial pemphigoid) |
LM: subepithelial separation DIF: linear band of IgG, C3 antibodies at basement membrane zone IIF: limited diagnostic utility |
| Lupus erythematosus | Malar rash Non-specific ulcerations OR lichenoid lesions with radiating striae SLE associated with multisystem complications |
LM: subepithelial and perivascular inflammatory infiltrate DIF: granular band of antibodies at basement membrane zone (lupus band test) Serum: elevated ANA (non-specific); anti-DS DNA, anti-Smith (highly specific) |
| Systemic sclerosis | Raynaud phenomenon Sclerodactyly, fingertip ulcerations Microstomia Widening of periodontal ligament space Loss of gingival attachment Resorption of ramus, condyle, coronoid process Fibrosis of skin and internal organs |
LM: accumulation of diffuse fibrotic collagen Serum: elevated ANA, anti-SCL 70 (non-specific) |
| Rheumatoid arthritis | Bilateral condylar flattening with irregular surface features Inflammatory destruction of joints |
Serum: elevated RF, ANA (non-specific); anti-ccp (highly specific) |
| Mixed connective tissue disease | Overlapping features of SLE, PSS, RA | Serum: elevated ANA (non-specific); anti-IU snRNP (highly specific) |
| Sjögren syndrome | Salivary hypofunction Dry eyes Joint pain Oral ulceration/pain Dental caries |
LM: periductal lymphocytic infiltrate and acini destruction Serum: elevated ANA (non-specific); anti-SSA, anti-SSB (highly specific) |
| Immune-Mediated Disease | ||
| Lichen planus | Wickham striae Desquamative gingivitis May become erosive, with shallow ulceration |
LM: presence of sawtooth rete pegs, subepithelial T-cell infiltrate DIF: subbasement membrane accumulation of fibrinogen |
| Recurrent aphthous stomatitis | Shallow round or ovoid ulcers with erythematous halo, on non-keratinized mucosa | LM: inflammation (non-specific) Serum: iron, vitamin B12, folate deficiency (associated very rarely) |
| Erythema multiforme | Target skin lesions Oral erosions exclude the gingiva Lip crusting |
LM: lymphocytes and histiocytes in superficial dermis, leukocyte exocytosis (non-specific) |
| Inflammatory bowel disease | Cobblestone, linear ulceration Pyostomatitis vegetans Aphthous ulcers Orofacial granulomatosis Angular cheilitis |
LM: neutrophil infiltrate in lamina propria, granulomatous inflammation DIF: immune reactivity to IgG, IgM, IgA |
| Orofacial granulomatosis | Recurrent facial swelling, including the lips and buccal mucosa | LM: edema in lamina propria, granulomatous inflammation (non-caseating), lymphocytic infiltrate |
| Wegener granulomatosis | ”Strawberry” gingivitis Oral ulceration |
LM: granulation tissue, eosinophils, multinucleated giant cells IIF: pANCA, cANCA (highly specific) Serum: elevated erythrocyte sedimentation rate, c-reactive protein (non-specific) |
| Sarcoidosis | Submucosal modules Bilateral parotid swelling Erythematous macules |
LM: epitheloid non-caseating granulomas Serum: elevated ACE, serum calcium (non-specific) |
| Allergic Disorders | ||
| Oral allergy syndrom/> | ||
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


