11
Congenital Syndromes with Dental Anomalies
11.1 Introduction
A syndrome is currently defined as the presence of multiple congenital developmental malformations, deformations and/or dysplasias, occurring in varying combinations with or without mental abnormalities, as a result of genetic aberrations or the effect of a teratogenic agent.
Congenital anomalies can be distinguished into:
- Malformations: morphological defects due to intrinsic disturbances of development
- Disruptive defects: developmental malformation due to an extrinsic cause
- Deformations: when abnormal mechanical or functional forces, such as lack of fetal movement, affect morphology, frequently in late fetal life (some disruptions cause secondary deformations)
- Dysplasias: morphological defects due to abnormal cell organization or function in a specific tissue.55 114
A “true” syndrome is due to a single etiological cause leading disturbances with primary effects in several tissues. In “false syndromes” several anomalies are present and they are caused by more than one etiological factor.51 Use of the term “syndrome” is incorrect in anomalies such as “crack(ed) tooth syndrome” and “Riga–Fedes syndrome”.176 A “sequence” is a primary structural abnormality, which through secondary and tertiary consequences results in a pattern or combination of developmental defects. Syndromes and sequences are named after their first and/or most important reporter or the main affected body part(s) or are referred to by the number (and part) of the chromosome responsible for the defects.51 52
11.1.1 Causes of Syndromes
- In many syndromes, the cause is a single mutated gene. Thousands of such monogenetic syndromes exist, and new ones are described every week. Mutations in several genes are associated with craniofacial and dental aberrations.328 Spontaneous mutations, which may become inheritable, occur, for instance, in more than half of tuberous sclerosis patients.106
- Other syndromes are caused by abnormalities in chromosome number. The germ cell or the female gamete may contain a pair instead of one chromosome; after fertilisation, three chromosomes instead of two will be present in the somatic cells. When only a part of a chromosome pair splits, the fertilised egg has two chromosomes plus that part. A (micro-) part of a chromosome may be absent: after fertilisation, the chromosome pair will be incomplete.
- Embryonic malformation may also occur due to the effects of exogenous teratogenic agents. Examples are: alcohol abuse, antenatal infections (congenital syphilis), metabolic disturbances and use of teratogenic medications such as thalidomide. The dosage, vulnerability of the mother/fetus, time of exposure and interactions between different agents determine the severity of the disturbances.106 Alcohol abuse by the expectant mother is estimated to affect 2 : 1000 newborns in the USA and the risk increases when alcohol is used together with tobacco.299
- Syndromes also result from a combination of genetic and exogenous causes (e.g. clefts).114
11.1.2 Causes of Sequences
The diagnostic determinants of the most frequent sequence, the Pierre Robin sequence (1 : 2000–30 000 births, Europe, ∼9 : 100 000 births) develop in the 10th–12th week in utero. Pathognomonic features are: mandibular micrognathia, cleft palate and glossoptosis (falling back of the tongue leading to airway obstruction). The causes of this sequence include:6 52 106 302
- Owing to insufficient amniotic fluid, the fetal mandible is pressed against the sternum leading to deformation. The tongue is located between the unfused palatal processes and prevents their fusion.
- Inadequate mandibular movements due to neurogenic hypotonia.
- Syndromes, including isolated cleft palate.
- Teratogenic agents.
- Collagen disturbances.
Severe airway obstruction and prolonged hypoxia further lead to cor pulmonale, brain damage, feeding problems, (lethal) apnoea,161 etc. Procedures such as intubation are used to bring the mandible forwards and to prevent glossoptosis. An orthodontic appliance that stimulates the vomiting reflex may help teach the patient to avoid responding to the reflex, by holding the mandible in a forward position.272
11.1.3 Genotype and Phenotype
In a number of syndromes, early ossification of the cranial sutures inhibits the growth of the skull (“craniosynostosis” with sometimes high intracranial pressure) due to mutation of a fibroblast growth factor receptor.7 56 121 There are a limited number of clinical manifestations of abnormal development. Thus, craniosynostosis, dwarf stature, polydactyly (too many fingers), microcephaly (small head) and micrognathia, to name a few, are not syndrome-specific, and are pathogenetically heterogeneous.52 121 In contrast, a mutated gene may lead to markedly varied phenotypes.99
11.1.4 Diagnosis
The overall pattern of anomalies, the mode of inheritance and the molecular basis (if known), all help in reaching a diagnosis, but the clinical diagnosis of a syndrome with non-specific and variable symptoms may be very difficult. Syndromes with a known aetiology are distinguished by the frequency and in what combination certain characteristic anomalies occur.
Immediately after birth, the head and neck may be deformed because of intra-uterine constriction, but this corrects spontaneously within a few days. If it does not, further examination must be carried out to check whether the deformation represents a true malformation. An example of late spontaneous disappearance is dolichocephaly (long-headedness) in prematurely born children. The heavy head and weak neck muscles force the child to lie on either the right or left side; the head becomes long and narrow because the soft and thin bones deform. After about 3 months, the dolichocephalic pattern disappears because the child increasingly moves his or her head. Dolichocephaly persists, however, in full-term children with premature closure of the side sutures in the skull.130
Many variations exist in the size and form of facial features such as the forehead, face, ears, distance between the eyes, nose and nostrils, lips and mouth (palate, uvula), etc., which can be measured and compared with normal values,101 but often one has to rely on subjective assessments.54 A single minor craniofacial deviation is in general no more than a family characteristic. Three minor deviations are associated with a major defect in more than half of the children;245 these deviations may be statistically uncommon in the population overall, yet may be part of normal familial morphological variants.51 However, a combination of minor facial anomalies may result in a specific appearance, such as in trisomy 21 and through their recognition lead to the diagnosis.54
The history, including pregnancy and birth history, contact with teratogenic and mutagenic agents, presence of dysmorphological features in other identically affected family members and a careful clinical examination may point to a diagnostic hypothesis. Additional genetic and molecular tests may confirm the diagnosis, which is a prerequisite for determining the prognosis, therapy (if possible) and need for genetic counselling.114 The general dental practitioner will encounter common syndromes such as clefts and trisomy 21, but it may be rare to see other uncommon ones. Thus the dentist must have some knowledge of the common syndromes for making appropriate referrals and arranging consultation and collaboration with geneticists and paediatricians/physicians.
11.1.5 Dental Anomalies
Either higher or lower frequency of occurrence of a dental anomaly in patients with a syndrome compared with the general population makes it likely that the dental anomaly is associated with the syndrome.51 Hyperdontia, double teeth, taurodontism, enamel hypoplasia, etc. are sometimes reported to occur as a part of a rare syndrome,118 139 275 but their presence may simply be coincidental (see Chapter 2 for detailed description of deviations of tooth number and shape). Isolated dental anomalies, such as hypodontia, may be micro-manifestations of a syndrome. For example, the combination of 10 agenetic maxillary posterior teeth, partial absence of the alveolar process and otitis media227 may or may not represent an as-yet unknown syndrome, but a deaf-mute patient with 14 congenitally missing teeth does not,240 because oligodontia may occur in some deaf-mute persons.
11.1.6 Nomenclature
The following terms are frequently used in relation to syndromic features.
- Brachy: short, such as brachycephaly (short head) and brachydactyly (short fingers).
- Chondros: cartilage (dyschondroplasia, achondroplasia = disturbed development of the cartilage).
- Dyshidrosis: disturbed function or absence of the sweat glands. Anhidrosis, hypohidrosis- and hyperhidrosis refer to, respectively, the absence, under- and over-production of sweat.
- Dysostosis: disturbed bone development, in particular disturbed ossification.
- Frontal bossing: prominent, pronounced forehead (brow).
- Hypertelorism: larger than normal distance, often pertaining to the eyes.
- Hypogenitalism: underdeveloped function/underdevelopment of the gonads.
- Onychodysplasia: abnormal nails.
- -ploid: a suffix indicating number of chromosomes. Haploid means 23 chromosomes, as normally present in the gametes. Diploid refers to the 23 chromosome pairs in the somatic cells. Aneuploidy indicates both fewer and more chromosomes than the haploid number.
- Poly: presence of extra elements in a body part: polydactyly (more than five fingers).
- Strabismus: cross-eyed.
- Syn: together, syndactyly (fused fingers or toes), craniosynostosis (early ossification of the sutures of the skull, sometimes one or more).
- Trichodysplasia: abnormal hair.
11.1.7 Classification
Of the monogenetic syndromes, more than 1000 manifest in the orofacial region. Most chromosomal syndromes have orofacial effects as do many syndromes with a multifactorial cause.275 Included here are only those monogenetic syndromes that include dental anomalies, unless they are very rare (arbitrarily considered as an prevalence rate of about 1 or <1 : 100 000, with one or two exceptions).
Extensive, well-documented descriptions of syndromes may be found in several textbooks,100 268 275 276 in particular in the lavishly illustrated, encyclopaedic Syndromes of the Head and Neck by Gorlin et al.106 The web-based “OMIM database” developed by McKusick et al. contains exhaustive descriptions of syndromes, grouped by the mode of inheritance and then alphabetically within the groups.211 Each syndrome has a six-digit identification number, unless allelic variants are known (when it has 10 digits). Other classifications also exist.53 In this chapter, McKusick’s classification is partly followed. Autosomal dominant (AD) syndromes are described in Section 11.2, which also includes the autosomal recessive (AR) and X-linked syndromes with identical names. Section 11.3 describes the AR syndromes, Section 11.4 the X-linked syndromes, and Section 11.5 the syndromes due to chromosomal abnormalities. Prevalence figures for Europe are based upon the “Prevalence of rare diseases: a bibliographic survey, September 2006” (last updated 2011; Orphanet Reports Series, http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_decreasing_prevalence_or_cases.pdf). For a few syndromes, only the number of cases is given.
11.2 Autosomal Dominant Syndromes
100800. Achondroplasia, Chromosome 4
Prevalence: 1 : 16 000–32 000.106
Bone growth impairment causes short-limb dwarfism (120–130 cm) with a normal torso. Striking features are an enlarged head (hydrocephalus internus), frontal bossing, depressed nasal bridge, midfacial dysplasia, mandibular protrusion, short neck, lordosis (abnormally curved spine), joint hypermobility but limitation of elbow extension, and trident (three pronged) hands.106 211
Anomalies in the teeth are seldom described.38 Besides taurodontism the incisor crowns may converge towards the incisal edge, with prominent mamelons suggestive of fusion with supernumerary or supplemental teeth.
101200. Acrocephalosyndactyly Type I, (Apert’s (Crouzon’s) Syndrome)
Prevalence: 1 : 65 000–100 000 births.55 106
This syndrome, which is one of the syndromes featuring craniosynostosis, is often due to a fresh mutation. Many affected babies die in the neonatal period.137 The premature fusion of skull sutures prevents the growth-related anterior displacement of the cranial base, orbits and maxilla. Late closure of the fontanelles underlies “acrocephaly” (tower skull) and “acrobrachycephaly” (short skull).
Facial characteristics include a steep and high forehead, hypertelorism and exophthalmia (bulging eyes), underdeveloped midface with a beaked nose and mandibular prognathism, abnormally shaped ears with hearing problems, symmetrical syndactyly and often mental retardation, but rarely an IQ = 35 has been reported.106 211 The high-arched palate is narrow, with lateral bony swellings, and sometimes present a cleft,244 ending in a bifid uvula.211 Other dental anomalies that have been reported are malocclusions (crowding), hypodontia,249 322 hyperdontia,106 delayed eruption,268 278 323 root resorption and transposition of teeth.323
Craniofacial measurements in Apert’s and Crouzon’s syndromes (see 101 200) deviate from the normal ranges, in general more so in Apert’s than Crouzon’s.156 After infancy, the differences become less exaggerated.158
113650. Branchio-Oto-Renal Dysplasia (Bor’s Syndrome), Chromosome 8
Prevalence: 1 : 40 000.221
There are fistulas or cysts in the lower neck, ear malformations, hearing loss and renal anomalies.106 211 221 Oral findings include an arched cleft palate, bifid uvula and retrognathia,106 211 generalised microdontia (rare in the permanent dentition) and malformed premolars.221
118400. Cherubism (Familial Benign Giant Cell Tumour of the Jaw), Chromosome 4
Prevalence: about 200 patients, twice more likely in males than females.
In this non-familial, inherited syndrome there is painless fibrous cyst-like dysplasia (central giant cell granuloma with failure of osteoclastogenesis).355 This results in overdevelopment of the jaws, starting at age 3–4 years and sometimes earlier, leading to an “angelic” round face with broad cheeks and hypertelorism.211
The deciduous teeth exfoliate early. Displacement of permanent tooth germs may lead to ectopic impaction301 and resorption. Oligodontia is also reported. After puberty, there is partial remission of cherubism. Surgical reconstruction should be delayed until the end of puberty,107 318 and ranges from tooth removal to correction of the shape of buccal bone, removal of giant cell tumour and autotransplantation of teeth to normal bone areas.301 The grade of severity of cherubism is related to the number of agenetic or impacted molars.301
Cherubism may be present in Noonan’s syndrome (Section 11.5) and also occur in combination with fibromatoses.106
Orofacial Clefts
119530. Orofacial Cleft 1, Chromosome 6
602966. Orofacial Cleft 2, (AD?), Chromosome 2
600757. Orofacial Cleft 3, (AD?), Chromosome 19
See also: 119300 and 119540
Prevalence: varies by country.
In the Netherlands, about 2.9 : 1000 children are born every year with orofacial clefts. Boys are affected more often than girls,44 84 290 and 0.4 : 1000 have a cleft palate (see also 119540).291 In Sweden, the incidence is double that in France and California, when patients from Asian ethnic backgrounds are excluded.237 All kinds of clefts are present in 0.2–3.6 : 1000 of newborns worldwide (although racial variation is there: Indians > Mongoloid > Caucasian > African).84 106 The relationship with race suggests an association with the facial or palatal width. More severe defects are generally noted in boys, although not in all races.106 Orofacial clefts should be distinguished from the isolated clefts of the palate and from the cleft of the lip and palate.
Cleft lip and/or palate present in 200–250 syndromes.106 211 In about 15% of cases, orofacial clefts occur in combination with other anomalies, such as the Van der Woude syndrome (119300) and chromosomal syndromes such as the trisomies.141 237 291Isolated clefts occur in 75–80% of cases, 10–15% are familial and about 15% occur as parts of a syndrome. If one parent and one child have a cleft, there is a 17% probability that a subsequent child will be affected.211 Neither higher maternal age nor higher birth order or paternal age is associated with the risk of isolated clefts.297 Environmental influences, including maternal drug and alcohol intake and smoking, are of importance.
In a sample of monozygotic twins, in about a third of cases, both twins exhibited orofacial clefts and a palatal cleft was present in one-quarter of both twins.121 Most dizygotic twins in another sample were discordant.205 A gene on the short arm of chromosome 6 has been implicated in clefts, but (modifying) effects of genes on chromosomes 4, 17 and 19 are also possible.121 Orofacial cleft 2 is linked to chromosome 2.211 In regards to race and environment, different transcription factors (such as transforming growth factors) regulating skull development are possibly involved, but which ones are is not known.165
Facial and cranial clefts are caused by non-fusion of embryonic parts (from the 7th embryonic week). The clefts are classified from 0 to 14.275 Cranial clefts above the eye are numbered 8–14 and facial clefts occurring under the eyes 0–7 (from the nose to the ear). Clefts of the lip, alveolar bone and palate are included in general in clefts 1, 2 and 3.
The severity of cleft lip, unilateral or bilateral, varies from a small notch in the lip to an actual cleft involving the alveolus and premaxilla. The palatal cleft manifests as a shallow groove in the midline of the hard palate or a full cleft through the bone. Unlike other clefts, palatal clefts are more prevalent in females than males and more frequently on the left than the right side. Two years after birth, development (weight) lags behind in children with cleft palates, and is attributed to feeding problems, airway infections and surgery.84 138 The child’s ability to learn to speak is hampered.
“Micro-clefts” in the middle of the lower lip are present in a small proportion of children with orofacial clefts and the Pierre Robin sequence.225
Solitary hypodontia is common in orofacial clefts;112 70–80% prevalence is reported.44 The findings related to hypodontia may be summarised as follows:
- Hypodontia is most frequent in the maxilla.228 The maxillary lateral incisor is most frequently agenetic, also when there is only a cleft lip (Figure 11.1).
- Hypodontia in the deciduous dentition is more often associated with non-alveolar clefts than in controls.63 The permanent lateral incisor in the cleft region is agenetic considerably more often than the deciduous incisor; when the latter is absent, its successor will be agenetic.296 Van der Wal reported that the lateral incisor was absent in half of one sample of patients with facial clefts and was malformed in one-quarter of the patients.292 The profile shows a retrusive upper lip/jaw.175
- The more severe the cleft, the more severe is the hypodontia,116 228 including the parts of the dental arches beyond the cleft region.6 228
- The upper central incisor is agenetic in small proportion of patients.296 The second maxillary and mandibular premolars are frequently agenetic in similar proportions312 (18% of the patients), and this is more on the left than the right side, where the clefts are more common.258
- The second molars are missing in about 4% (3% of contralateral teeth develop late).116
- Hypodontia is present to the same degree in familial and sporadic clefts.44
- The development of the dentition is delayed.116 270
Figure 11.1 Patients with clefts of the lip (A) and palate (B).
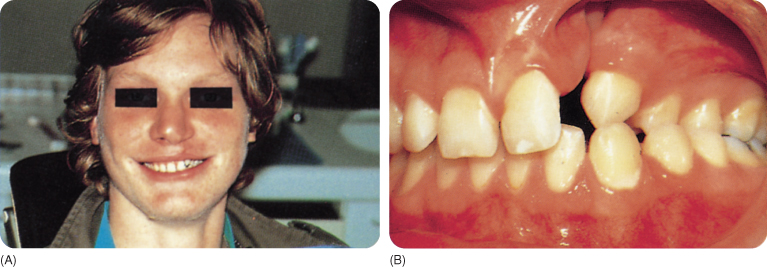
The deciduous teeth are relatively small, yet crowding is almost the rule.63 The permanent teeth have normal measurements.215 Tooth development and eruption are delayed in particular in cases with hypodontia and worsens with age.224 228 The morphology of, in particular, the upper lateral incisor is affected;296 T-shaped incisors have been reported.113 155 The enamel of the central incisor is more often hypoplastic than that of the lateral incisor,296 as found in 20–45% of one series of cases,109 perhaps as a consequence of surgery,3 although this also occurs in non-surgical patients. Enamel opacities (of which many are carious white spots) are present in 95% of 5–17-year-olds with clefts.109 Caries develops 3.5 times more often than in controls and is not restricted to the teeth near the cleft.31 Intranasal teeth have been reported a few times, attributed to the presence of clefts and surgery.147 Small morphological tooth deviations have been described in the region outside the cleft.35 Lip and palate clefts are regularly accompanied by hyperdontia, also in combination with hypodontia and natal and neonatal teeth,5 44 63 126 155 197 207 229 236 296 309 outside the cleft region.
In about 20% of cleft cases, there are extra lateral incisors and fewer extra central incisors in both dentitions.296 In about 25% of the deciduous and about 10% of the permanent dentitions supplemental lateral incisors are present,5 on average about 0.3 extra teeth/dentition,44 155 in particular when the lip is involved in an alveolar cleft.308
Early removal of (impacted) extra teeth in the cleft results in unfavourable loss of bone; the tooth must be removed during secondary bone plastic surgery, at the age of 7–12 years.92
Other Syndromes with Clefts
119300. Van Der Woude Syndrome, Chromosome 1
607613. Van Der Woude Syndrome 2, Chromosome 1
Prevalence: 1 : 35 000–100 000.106
The “lower lip sinus syndrome” is attributed to absence of a part of chromosome 1 (or 2). The majority shows symmetrical pits and/or sinuses (pits connected to small salivary glands) in the lower lip. The penetrance is high and the expressivity is variable.42 Sometimes slight bulges are present and less often a cleft lip and/or palate and/or uvula, frequently with hypodontia.211 231
119540. Cleft Palate, Chromosome 2
303400. Cleft Palate with Ankyloglossia, X-Linked
Prevalence: 0.4 : 1000.106
Non-familial cleft palates seem related to environmental factors (maternal age).211 Clefts of only the palate are more common in females.5 (Submucosal) palatal clefts occur in association with other anomalies more often than orofacial clefts.106 Half of 139 patients with cleft palates had multiple anomalies, and 34, in particular, had Stickler’s syndrome: a flat midface, joint problems, hearing loss and myopia (short-sightedness).141 Additional problems, e.g. the Pierre Robin sequence (261800) and the Van der Woude syndrome (119300), were present in more than half of another sample of 50 cleft palate patients.291 Associations with Apert’s (101200) and the de Lange (122470) syndromes are also reported.143
Agenetic maxillary lateral incisors (more frequently absent in the Pierre Robin sequence)6 and enamel hypoplasia of the central incisors are associated dental anomalies. Anterior tooth agenesis is less frequent than in patients with orofacial clefts, but has been noted to occur more often than in controls.5
Ranta et al. found that children with hypodontia had similar proportions of 1–11 agenetic teeth as children with cleft palates, but in the latter a larger number of maxillary teeth were absent. More teeth were symmetrically agenetic in the maxilla of children without a cleft and in the mandible of children with a cleft.230
In about 15% of patients, a submucous cleft palate is associated with isolated dental agenesis, mainly the mandibular second premolars followed by the maxillary lateral incisors and second premolars. Heliovaara et al. reported peg-shaped lateral incisors in 10%, transposition (Mx.C.P1) in 4% and supernumerary teeth in 3% of their sample of cleft palate patients.120
In cleft palate patients, odontogenesis is delayed, and more so when there is associated hypodontia.226
119600. Cleidocranial Dysplasia (Formerly Dysostosis Cleidocranialis), Chromosome 6
Prevalence: 1 : 1 000 000.256
The syndrome is caused by mesodermal dysfunction due to anomalous actions of osteoclasts, resulting in multiple bone anomalies. About a third are due to spontaneous mutation.106 The cranial sutures are wide open, and some of them and the frontal fontanelles do not ossify (requiring autologous bone transplantation). The features include brachycephaly, frontal bossing, hypertelorism, depressed nose bridge, hypoplastic zygomas and maxillae, hypoplastic frontal and paranasal sinuses and a small midface. The patients have drooping, narrow shoulders and a long neck, with short stature. The clavicles may be (partially) absent, which allows the affected individual to bring the shoulders close together (Figure 11.2).34 106 211
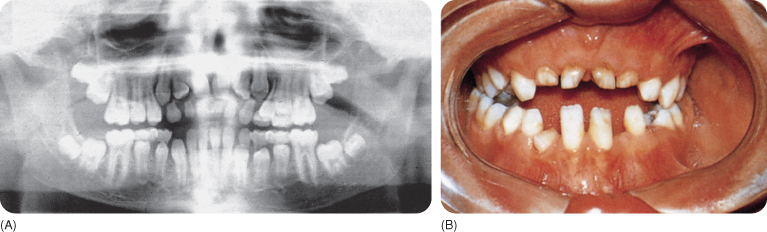
The jaw bones may show coarse trabeculation and the alveolar crestal bone may be very dense.181 The underdeveloped maxilla makes the mandible appear more prominent.8 115 There is a high palatal vault101 and occasionally a cleft, which may or may not be submucous. The permanent dentition may have a few to more than 30 extra teeth,8 13 24 47 137 324 mainly in the mandibular premolar region,144 which develop after the age of 12 years,181 and in the premaxilla.12 88 137 These teeth may develop from remnants of the dental lamina.157 The permanent teeth mature late, which is more evident when accompanied by hyperdontia.256 One sample of siblings showed discordance in the location and numbers of extra teeth.325 Rarely, a tooth from the normal series is agenetic.24 65
The eruption of the permanent teeth is delayed (Figure 11.3) or fails65 (partially in some incisors),24 except for the permanent first molars. The deciduous teeth resorb late and persist into adulthood.8 9 144 245 256 311 Cellular cementum is absent and acellular cementum is grossly lacking on the impacted teeth.254 A number of teeth, in particular the first molars,137 erupt in spite of the absence of cellular cementum.258 267 The eruption delay seems to be caused by the reduced resorption of bone and roots of the deciduous teeth.106 157 244 The roots of the unerupted teeth are (almost) completely developed, though they may be curved,144 long or short, with a sharply pointed apex or malformed otherwise.181 The crowns are often small.
Correction of the underdeveloped midface is achieved with surgery.89 Surgical intervention enables the eruption of impacted teeth, with formation of cementum,122 but in some patients orthodontic traction is desirable.65 Stepwise early removal of the extra and persisting deciduous teeth, along with the overlying bone, is followed by spontaneous eruption of the normal teeth.137 Other authors state that the bone must be saved as much as possible: the permanent successor is extruded through an opening in the bone after extraction of the deciduous tooth.24
Presence of different mutations was established in two patients belonging to the same family and in another patient from a different family.99 315 The mutations concern the gene CBFA1. In an experimental study, homozygous mice did not develop a skeletal system and heterozygous mice had cleidocranial dysplasia without the dental anomalies.315
Figure 11.2 Cleidocranial dysplasia: the shoulders can be brought much closer together than in normal individuals.
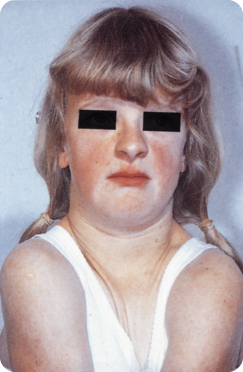
Figure 11.3 (A,B) The dentition of a patient with cleidocranial dysplasia; this case does not demonstrate hyperdontia.
122470. Cornelia De Lange Syndrome (Brachmann–De Lange Syndrome; Typus Degenerativus Amstelodamensis), Chromosome 3
Prevalence: 0.6 : 100 000 (Denmark),137 1 : 16 744 (Taipei) and 1 : 10 000 births (USA).106
Most cases are isolated.211 The syndrome, characterised by a delay in growth and development, has a mild, classical expression. The patients have low birthweight, short stature and micro-brachycephaly. The characteristic facial features are: hirsutism (with hair growth extending down to the neck and covering the forehead), approximating eyebrows above a depressed nose bridge and long eyelashes, anteverted nostrils with a long philtrum and an upward tilt of the nose tip, and maxillary prognathism with a thin upper lip and drooping oral commissures. The eyes show several anomalies. The characteristic facies does not develop until the age of 2 years in the mild phenotype and is present at birth in the classical type. The low-set ears may be small and curly hair may be present, for instance, on the back. The extremities in particular are also malformed, with oligodactyly and syndactyly of the second and third toes. Other abnormalities concern the gastrointestinal and genitourinary tract, such as hypospadias (ectopic external urethral orifice), hypoplastic kidneys and cardiovascular defects. Severe to borderline mental retardation exists.211
As regards the dentition, patients may show deciduous double teeth,72 delayed eruption, hypodontia and microdontia326 75 with spaced teeth.211 Gastric reflux causes dental erosion.326
123500. Crouzon’s (or Morbus) Syndrome, Chromosome 10
Prevalence: 1 : 25 000.106
A third of craniofacial dysostosis/pseudo-Crouzon’s syndrome cases211 256 are caused by fresh mutations. The growing skull is displaced downwards under pressure from the growing brain in one or another direction, depending on which sutures close early.235 The face is often broadest at the level of the eyebrows.206 Abnormalities in the vicinity of the eyes are the most frequent,153 and hearing deficits are also frequent,106 262 including hypertelorism and exophthalmia; blindness is not uncommon. The midface, including the maxillary arch, is hypoplastic (Figure 11.4). The nose may be beak shaped, above a short upper lip.211 Early surgery90 is done to reopen the sutures; screws are inserted to provide traction that promotes growth of the skull in the desired directions. The hands (and feet) exhibit syndactyly, brachydactyly and other defects.7 262
Figure 11.4 Underdeveloped midface in Crouzon’s syndrome.
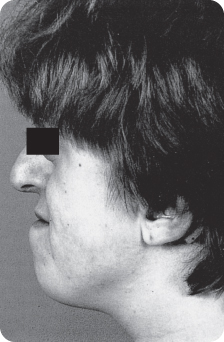
The short maxilla has a high palate, with lateral bony swellings. The associated malocclusions include crowding, open bite, cross bites, and a Class III molar relationship. Other dental anomalies that have been reported are hypodontia and small crowns,208 shovel-shaped incisors106 and impacted teeth.
129200–129550. The Ectodermal Dysplasias
Prevalence: (all types): 7 : 10 000.
The ectodermal dysplasias are a heterogeneous group of congenital disorders in which structures of ectodermal origin exhibit dystrophy; however, non-ectodermal tissues may also be affected.164 The main characteristic features concern the hair, teeth, nails and sweat glands:217
- Hypotrichosis: sparse, thin and sometimes dry hair
- Oligodontia
- Onychodysplasia (dystrophic nails)
- Dyshidrosis: affected sweat glands may be anhidrotic, hypohidrotic (in 80%) or hidrotic.
Salivary production is also reduced. Perspiration is possible to a degree in those with hypohidrosis; anhidrotic patients are in fact hypohidrotic. Consequences include pyrexia of unknown origin in children and excessive rise in body temperature in hot weather (which can cause brain damage that is sometimes fatal) and even after mild exercise. In a group of patients with various forms of ectodermal dysplasia, the number of aplastic teeth ranged from 2 to 26; all the third molars were missing. The teeth least affected were the maxillary central incisors and the mandibular canines, (which were, however, most affected by malformation) and the mandibular first molars.327
Two of the four characteristic features must be present in order to diagnose ectodermal dysplasia; the validity of the diagnosis increases when the triad of hypohidrosis, hypotrichosis and oligodontia is present. Moreover, patients often have:
- Asteatosis (reduced production by the sebaceous glands)
- A thin, smooth skin
- Full lips.
Other features include a small face, depressed nose bridge (saddle nose) with frontal bossing, absence of or more than two nipples, aplastic or hypoplastic mammary glands, patchy distribution of body hair, mental retardation106 288 and lacrimal gland hypoplasia. In the Christ–Siemens–Touraine syndrome (the most common form; see below) patients have short stature211 and cranial abnormalities (smaller face because of frontal bossing). Clefts, syndactyly, polydactyly241 and other features, such as deafness, absent dermal ridges or immunodeficiency with osteoporosis may also be present.211 The (oral) mucosa may be affected,98 and xerostomia may be present.260
Pinheiro et al. in 1981 described 18 types of ectodermal dysplasia, each of which included at least two characteristic features.217
Later, other forms were described, for instance odonto-onycho-dermal dysplasia (with features such as hidrotic palmoplantar hyperkeratosis, persisting deciduous teeth, severe hypodontia, dystrophic nails, erythema of cheeks/nose)81 183 186 and a variant with metal retardation and marked oligodontia of the permanent dentititon.187 More than 150 ectodermal dysplasias have been recognised.164
Presently, more than 200 types363 are classified according to all possible combinations of the four main features. When only two of the four principal defects are present, with or without other malformations, the syndrome belongs to group A. If only one of the four principal characteristics is present with at least one other ectodermal defect, the syndrome belongs to group B. Eleven subgroups are distinguished, based on the combinations of the presence of the principal characteristics, for instance defects of hair and teet/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


