Introduction
Nonsyndromic primary failure of eruption (PFE) is a rare autosomal dominant disorder of dental eruption with no obvious dental or soft tissue interference. The purposes of this study were to genetically and clinically characterize a family with many members affected by PFE and to describe the natural evolution of the disorder.
Methods
Three generations of a family with 18 members, 10 of them clinically affected by PFE, were evaluated periodically during 20 years of clinical follow-up. PFE was observed in varying degrees of severity in both sexes. Clinical presentation became more severe in adulthood. One patient had spontaneous reeruption of 2 posterior teeth. Cervical root resorptions were observed in 3 members. Genetic analysis showed a deleterious heterozygous mutation in intron 9 of the PTH1R gene (c.639-2A>G) and diagnosed an additional affected member.
Conclusions
The long-term follow-up of PFE cases in this family permitted the following observations: (1) the onset occurred from the preemergence to the postemergence phases, (2) PFE appeared to be closely related to ankylosis, (3) affected teeth maintained the eruptive potential even in adulthood, (4) the earlier the onset the more severe the open bite, and (5) cervical root resorptions occurred in 3 affected members.
Highlights
- •
Onset of primary failure of eruption (PFE) occurred from the preemergence to postemergence phases.
- •
PFE is closely related to ankylosis.
- •
Affected teeth maintained their eruptive potential even in adulthood.
- •
The earlier the onset, the more severe the open bite.
- •
Cervical root resorption occurred in some affected members.
Nonsyndromic primary failure of eruption (PFE) is a term used to describe a rare disorder in the dental eruption process with no obvious dental or soft tissue interference or associated systemic or syndromic conditions. Although few and isolated cases were reported before 1981, the first definition of the disorder can be attributed to Proffit and Vig. They originally described the condition as a cessation of the eruptive process that affects posterior teeth more severely, rarely in a symmetric pattern. Anterior teeth almost never are affected. Involved teeth may initially respond to orthodontic forces, but ankylosis occurs invariably. In other words, affected teeth seem to fail in the eruption propulsive movement and are not believed to be ankylosed.
The condition was further subclassified into type I, characterized by progressive open bite from the anterior toward the posterior segment, and type II, in which the second molar has some, although inadequate, eruption potential. The timing of type I onset is speculated to occur at the same time as craniofacial development of all affected teeth, whereas type II seems to be related to the stage of root development. Both types may coexist in different quadrants in the same patient, a condition that was further classified as type III.
Although some authors have been reluctant to speculate about the genetic etiology, recent studies have demonstrated heterozygous pathologic mutations in the parathyroid hormone receptor 1 (PTH1R) gene, with a mode of inheritance compatible with autosomal dominant transmission.
Differential diagnosis includes mechanical interference with eruption, syndromes associated with eruption defects, and long-term sequelae of radiotherapy. Mechanical interference caused by tongue or lip interpositioning leads to an open bite at nearly the same vertical level. Conversely, teeth lay at different vertical levels in PFE. When the eruption ceases after tooth emergence, it may also be diagnosed as secondary retention, which is attributed to ankylosis. Clinically, it is challenging to distinguish secondary retention from PFE.
Treatment of PFE depends on the degree of clinical severity but usually leads to frustrating results. In severe cases, orthodontic treatment, even associated with surgical procedures, has not resulted in effective establishment of occlusion. Prosthetic techniques are frequently the most feasible treatment option.
The rarity of PFE still leads to several gaps in our comprehensive knowledge of its pathogenesis. It is intriguing that a genetically determined condition can result in asymmetrical impairment of the eruptive movements and in various degrees of severity in different segments of the dental arches. Long-term clinical and radiographic studies on the natural course of PFE may provide the basis for determining the prognosis and provide new insights into the pathologic mechanisms. The aim of this study was to genetically and clinically characterize a family with 11 members affected by PFE in 20 years of follow-up.
Case report
This study was carried out in accordance to the guidelines of the Declaration of Helsinki and was approved by the ethics committee of the Dental School of Araçatuba (protocol 00839/2011) in Brazil. Written informed consent was obtained from all family members that permitted blood sampling for genetic analysis.
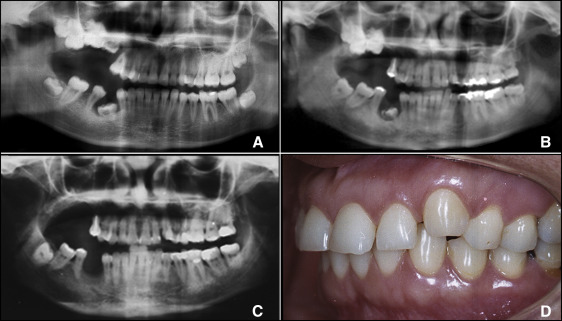
A 15-year-old girl (patient II.9) was referred to the Oral Surgery Department because of an infectious process caused by the maxillary deciduous first molar of the right side. The tooth was severely submerged, and its position had led to a periodontal pocket and an acute periodontal abscess. Clinically, a unilateral posterior open bite was observed at the right side. The panoramic radiograph showed inclusion of all maxillary permanent molars of the affected side, with no discernible mechanical interference ( Fig 1 , A ). The maxillary deciduous tooth was extracted. At the age of 23, the patient returned for extraction of the mandibular deciduous molar of the affected side due to suppuration via periodontal pocket. It was observed that the maxillary canine of the left side was slightly above the occlusal plane ( Fig 1 , B ). This infraocclusion was evident at the age of 42, when the mandibular right second molar was completely present in the oral cavity, and all maxillary molars of the same side had already been extracted. The maxillary second and third molars were fused by concrescence. A peculiar finding observed in the panoramic radiographs was the apparent increasing proximity of the mandibular right third molar to the mandibular channel during the follow-up period.
The family was investigated with a follow-up of 20 years, from the time when patient II.9 was 23 years old. Ten of 18 members were clinically affected with different degrees of severity, some of them clearly bilateral, with no difference between sexes ( Fig 2 ). Patients I.1 and I.3, as well as II.1, II.2, and II.7 were partially edentulous in the posterior segment; this did not permit any clinical conclusions concerning the diagnosis of PFE. Three members had anterior teeth in infraocclusion. Except for third molars, affected teeth had the alveolar bone crest at the level of the cementoenamel junction. There was no evidence of systemic disease in the affected people, and no other features of craniofacial and skeletal malformations or facial asymmetry. Dental arches were well developed, and diastemas were a common finding.
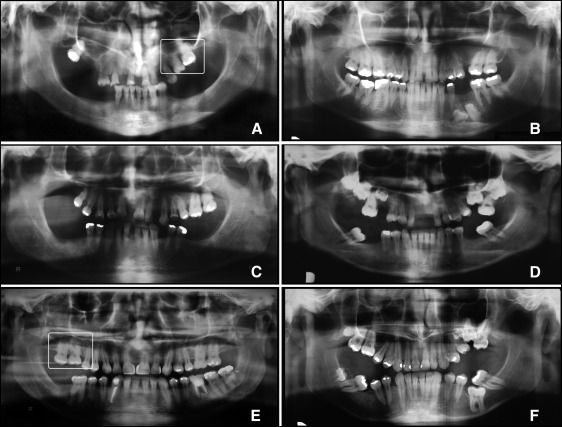
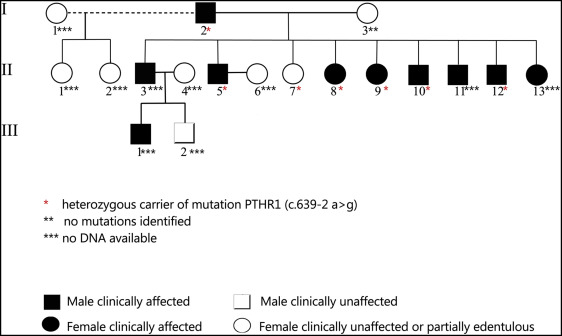
Figure 3 depicts the familial pedigree. During interviews, it was reported that 2 members of the third generation are affected by PFE additionally. However, they were not available for clinical and radiographic examinations and were not included in this report.
During the follow-up period, no treatment was carried out for open-bite correction, except for tooth extractions for periodontal reasons in several members of the family. Dental extractions did not cause any positional modification of adjacent teeth, in vertical direction or in mesial drift. In general, the condition worsened in adulthood, when in some cases PFE became prominent.
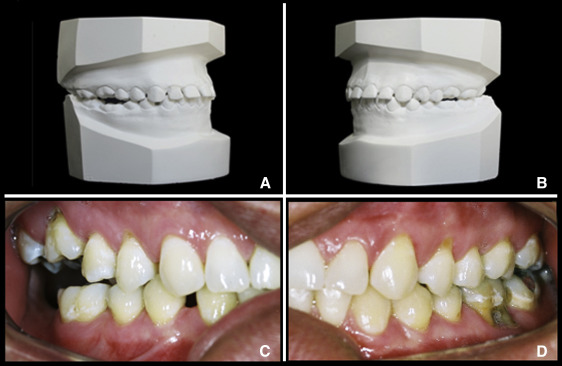
Patient II.11 had a peculiar course of development. He was unaware of his condition at age 18, since only the first molars were bilaterally in mild infraocclusion ( Fig 4 , A and B ). After 20 years, there was a clear open bite at the right side. Surprisingly, the first molars on the left side were in occlusion, and the mandibular right first molar was at the occlusal level. Additionally, the diastemas between the maxillary left and mandibular canines and first premolars were closed ( Fig 4 , C and D ).
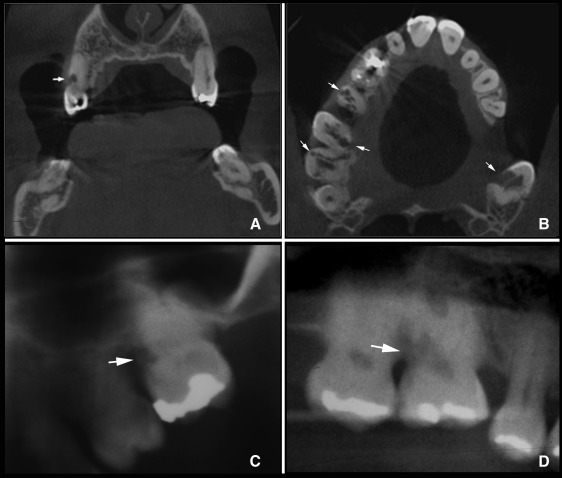
Multiple external cervical root resorptions involving the maxillary posterior teeth were diagnosed in patient II.12 at age 40. External root resorption was initially diagnosed in the maxillary right second premolar, which radiographically had a mottled aspect in an infrabony cavity. The patient reported unspecific and sporadic discomfort. The root canal seemed to be intact. A cone-beam computed tomography image showed further external root resorptions in the maxillary right first molar and the maxillary second molars of both sides ( Fig 5 , A and B ). Although these resorption lesions were invasive, they were asymptomatic. Radiographic images compatible with cervical root resorption were also found in the maxillary left first molar of patient I.2 ( Fig 5 , C ) and in the maxillary right first molar of patient II.11 ( Fig 5 , D ); all were in infraocclusion.
Genetic analysis
DNA analysis was carried out in the index patient II.9 by Polymerase Chain Reaction amplification and subsequent didesoxynucleotide sequencing of all coding exons and the immediate flanking exon/intron boundaries (±14 nucleotides) of the PTH1R gene (ref. seq. NM_000316.2 ), whereby “intron” is defined as a nucleotide sequence within the gene which is removed by RNA splicing during mRNA maturation, and an “exon” is a nucleotide sequence that is still part of the mature mRNA after introns have been removed. The primer sequences were described in a previous study. A heterozygous mutation (c.639-2 A>G) affecting the acceptor splice consensus sequence was found flanking exon 9 of the PTH1R gene. Split eukaryotic genes like PTH1R require the removal of the intronic sequences by the spliceosome and rely on highly conserved sequences at the borders between the respective intronic and exonic sequences. This is particularly true for mutations affecting 1 of the 2 nucleotides at the invariant. AG dinucleotides within the splice acceptor site present at the 3′ end of all known introns or the invariant GT dinucleotide within the splice donor site present at the 5′ end of nearly all known introns. Regarding c.639-2 A>G, this mutation affects the highly conserved adenosine nucleotide at position -2 at the splice acceptor site and thus renders the invariant AG dinucleotide ineffective for the splicing machinery. As a result, the mutation most likely leads to impairment in the mRNA splicing process and is expected to produce a nonfunctional receptor due to protein truncation. The pathogenicity of the mutation is predicted by the splice site prediction suite of the Alamut Mutation Interpretation Software (Interactive Biosoftware, Rouen, France), although the exact nature of the mutational consequences is unknown and would require the sequencing of the mutant transcript ideally in the affected dental tissues. Interestingly, at the same position, a different splice site mutation (c.639-2 A>C) was previously reported and was also predicted to result in an aberrantly spliced transcript. Both mutations are extremely rare and are not present in any of the 4 major public databases on human gene variations, including the dbSNP database (build44), the ExAC database , the NHLBI GO Exome Sequencing Project ( http://evs.gs.washington.edu/EVS/ ), and the 1000 Genomes Project.
To confirm segregation of c.639-2 A>G with disease in our pedigree, family members I.2, I.3, II.5, II.7, II.8, II.10, and II.12 were specifically sequenced for the absence or presence of the c.639-2 A>G mutation ( Fig 3 ). Clinically affected family members I.2, II.5, II.8, II.10, and II.12 all were heterozygous carriers of the splice site mutation. Patient II.7 also carried the heterozygous mutation c.639-2 A>G, although not clinically diagnosed because of the edentulous condition, giving a total of 11 affected members in this family. Family member I.3 showed the reference sequence.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


