Cornelia de Lange syndrome
- •
Syndrome is a clinical diagnosis.
- •
Many craniofacial abnormalities include synophrys, long philtrum, and cleft palate.
- •
Limb malformations are seen in these patients, along with toe fusion.
- •
Developmental delay is common.
Genetics
Multiple genes have been associated with this syndrome, including nipped-B-like protein (NIPBL) on chromosome 5, SMC1A, SMC3, and HDAC8 on the X chromosome. Most of these cases are secondary to spontaneous genetic mutations.
Clinical features
Cornelia de Lange syndrome is a clinical diagnosis, based on signs and symptoms. Certain features are noted, including low birth weight, developmental delay, small stature, microcephaly, low hairline with synophrys, flat nasal bridge, long philtrum, low-set ears, widely spaced teeth, cleft palate, limb differences, excessive body hair, and partial fusion of the second and third toes. Children often have long eyelashes ( Fig. 1 ).
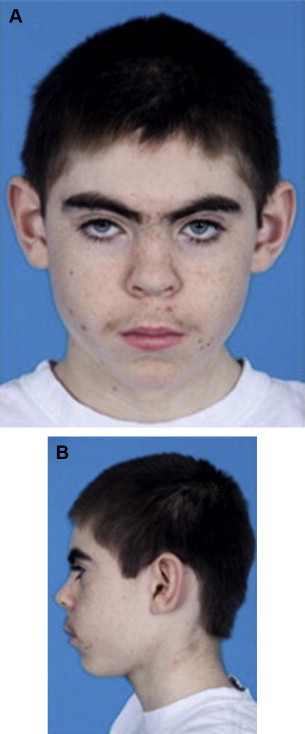
Cardiac abnormalities may also be present at birth. Gastrointestinal difficulties, including vomiting, diarrhea, and constipation, are common.
Behavior issues may play a role in children as well, including self-injury and aggression. Patients may display behaviors similar to autism.
-
Differential diagnosis
-
Whistling face syndrome
-
Fetal alcohol syndrome
Treatment considerations for Oral and Maxillofacial Surgeons
A thorough physical examination is important at birth to look for craniofacial abnormalities associated with this syndrome. A cleft palate, if present, should be noted, in order to plan for correction before speech development.
Because of the multisystem involvement of this syndrome, including congenital cardiac defects, it is important to consult the patient’s primary care physician before any procedure with or without conscious sedation.
Prader-Willi syndrome
- •
Most patients inherit disease from paternal chromosome 15.
- •
Patients have severe hypotonia and feeding difficulties in infancy.
- •
Patients develop morbid obesity and diabetes.
- •
Patients have hypogonadism with incomplete pubertal development.
- •
These patients show learning disabilities and behavioral problems.
Genetics
Prader-Willi syndrome is sporadically inherited from deletion or impaired expression of chromosome 15, specifically 15q11-13. Diagnosis is confirmed using DNA methylation or fluorescent in situ hybridization analysis. The incidence ranges from 1 in 10,000 to 1 in 20,000. There is equal distribution in male and female patients, as well as along differing ethnicities. In 70% of the cases, the abnormal gene is from a paternal inheritance; however, uniparental maternal disomy can lead to Prader-Willi syndrome in 25% of cases, with purely abnormal gene expression comprising 1% to 5% of the cases.
Clinical features
These patients typically show central infantile hypotonia with a poor suck at birth. The hypotonia is universal and can also lead to lethargy, a weak cry, and decreased movement. This condition leads to infant feeding difficulty, which causes an extreme drive to eat. Patients typically display morbid obesity, with short stature. The inability to feel satiated is secondary to hypothalamic abnormalities and causes patients to display food-seeking behavior, hoarding food, and eating of inedible objects. These patients develop sleep apnea and diabetes. It is common to see psychological disturbances, including obsessive-compulsive disorder (OCD) and oppositional behavior possibly secondary to hypothalamic dysregulation. Skin picking is a common manifestation of OCD. Patients with Prader-Willi have several dysmorphic features, including almond-shaped eyes; dolichocephaly; and small mouth, hands, and feet. The characteristic facial features may not be apparent at birth, but develop over time. Prosodic speech is common, secondary to a speech articulation defect. In addition, these patients show a global motor delay, as well as learning disabilities caused by a low intelligence quotient ( Fig. 2 ).
-
Differential diagnosis
-
Craniopharyngioma
-
Central nervous system depression
-
Neonatal sepsis
-
Myopathy
-
Fragile X syndrome
Treatment considerations for Oral and Maxillofacial Surgeons
These patients have numerous abnormalities, and are best served with a multidisciplinary approach. In oral and maxillofacial surgery, the patients are high risk when considering in-office surgical procedures and intravenous sedation. In addition to the behavior management skills required, attention needs to be directed toward airway management in these patients. Many are obese and produce a thick viscous saliva. Patients with Prader-Willi can also show apneic episodes and are hypotonic. A sleep study may be indicated in patients with Prader-Willi. These concerns need to be addressed before conscious sedation, and consideration of an operating room setting is advisable. Early use of growth hormone replacement in these patients can prevent the development of the facial dysmorphic features.
Prader-Willi syndrome
- •
Most patients inherit disease from paternal chromosome 15.
- •
Patients have severe hypotonia and feeding difficulties in infancy.
- •
Patients develop morbid obesity and diabetes.
- •
Patients have hypogonadism with incomplete pubertal development.
- •
These patients show learning disabilities and behavioral problems.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


