Key points
- •
Genetic alterations can cause physiologic responses resulting in systemic and metabolic complications.
- •
These complications can cause significant deficits, as in Hurler and Williams syndromes, which can lead to debilitating disease and early death.
- •
These alterations may also lead to abnormal immune responses that affect significant organs and organ systems.
Fabry disease
- •
Fabry disease, also known as Anderson-Fabry disease, is an inherited X-linked recessive disorder of lysosomal storage.
- •
Fabry disease is the second most prevalent lipid storage disease after Gaucher disease and occurs from a deficient activity of lysosome hydrolase, alpha-galactosidase A.
Genetics
The alpha-galactosidase A gene has been mapped to the region q22.1 of the X chromosome. When mutated, a progressive accumulation of a glycolipid called globotriaosylceramide (Gb3) affects mainly endothelial and vascular smooth muscle cells and causes damage to many major organs including renal and cardiac insufficiency, and central nervous system disorders including cerebral infarction and skin and pulmonary symptoms. It is mostly a male disease with the reported incidence varying from 1 in 17,000 to 1 in 117,000 men in white populations. However, this disease is probably unreported because many of the manifestations are nonspecific, the disease is rare, and often the initial diagnosis is not accurate. Women can have the disease but usually have milder symptoms that occur later in life. There are rare reports of women being asymptomatic early in life, but then showing symptoms as severe as those of men with the classic phenotype.
Clinical features
Clinical manifestations of the disease often occur early in life, caused by the increase in glycolipids, which causes inflammation and fibrosis. Therefore, severe neurologic pain in the limbs, especially during times of stress, begins around the age of 10 years. Renal dysfunction with proteinuria and polyuria, often leading to renal failure, is one of the serious complications of the disease. Skin lesions are often present, including hypohidrosis and angiokeratomas. The labial mucosa is often involved with a maculopapular eruption ( Fig. 1 ). Corneal changes are usually an early sign with a whorl-like keratopathy seen on a slit lamp examination ( Fig. 2 ). As these patients age, central nervous system involvement often leads to increased incidence of stroke. Cardiovascular effects include hypertension and cardiomyopathy with eventual heart failure. Oral and maxillofacial findings include increased prevalence of maxillary cysts and maxillary prognathism. Thickening of the lips and a bulbous nose have been described in many cases. Perioral linear telangiectases and cases of granulomatous cheilitis and granulomatous gingivitis are noted. Tinnitus is reported in approximately 40% of patients with this disorder. Because of the severe major organ dysfunction, most of these patients die at an early age secondary to stroke, end-stage renal failure, or cardiovascular complications.
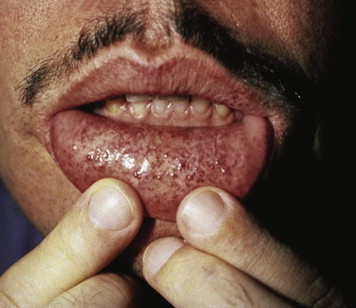
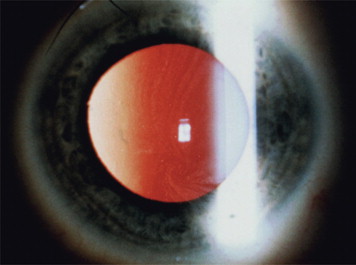
Differential diagnosis
Because this disorder is rare, it is often misdiagnosed because of early nonspecific manifestations, including neurologic and skin disorders. These disorders can include rheumatoid arthritis, fibromyalgia, multiple sclerosis, and Meniere disease. The disorder is often discovered following more serious complications such as stroke, cardiac insufficiency, and renal failure. Because treatment with enzyme replacement therapy has been shown to have positive results, early diagnosis of Fabry disease is important. When suspected, serum assays of leukocyte alpha-galactosidase A activity are effective in men; however, genetic testing in women is recommended because of inclusive serum results.
Treatment considerations for the oral and maxillofacial surgeon
Oral surgeons should be suspicious that any patient displaying angiokeratomas of the face, cheilitis granulomatosa, or granulomatous gingivitis may have the potential for Fabry disease, especially young men. With late-stage disease and renal, cardiac, and neurologic complications, treatment must be carefully administered with close consultation with the appropriate specialists.
Fabry disease
- •
Fabry disease, also known as Anderson-Fabry disease, is an inherited X-linked recessive disorder of lysosomal storage.
- •
Fabry disease is the second most prevalent lipid storage disease after Gaucher disease and occurs from a deficient activity of lysosome hydrolase, alpha-galactosidase A.
Genetics
The alpha-galactosidase A gene has been mapped to the region q22.1 of the X chromosome. When mutated, a progressive accumulation of a glycolipid called globotriaosylceramide (Gb3) affects mainly endothelial and vascular smooth muscle cells and causes damage to many major organs including renal and cardiac insufficiency, and central nervous system disorders including cerebral infarction and skin and pulmonary symptoms. It is mostly a male disease with the reported incidence varying from 1 in 17,000 to 1 in 117,000 men in white populations. However, this disease is probably unreported because many of the manifestations are nonspecific, the disease is rare, and often the initial diagnosis is not accurate. Women can have the disease but usually have milder symptoms that occur later in life. There are rare reports of women being asymptomatic early in life, but then showing symptoms as severe as those of men with the classic phenotype.
Clinical features
Clinical manifestations of the disease often occur early in life, caused by the increase in glycolipids, which causes inflammation and fibrosis. Therefore, severe neurologic pain in the limbs, especially during times of stress, begins around the age of 10 years. Renal dysfunction with proteinuria and polyuria, often leading to renal failure, is one of the serious complications of the disease. Skin lesions are often present, including hypohidrosis and angiokeratomas. The labial mucosa is often involved with a maculopapular eruption ( Fig. 1 ). Corneal changes are usually an early sign with a whorl-like keratopathy seen on a slit lamp examination ( Fig. 2 ). As these patients age, central nervous system involvement often leads to increased incidence of stroke. Cardiovascular effects include hypertension and cardiomyopathy with eventual heart failure. Oral and maxillofacial findings include increased prevalence of maxillary cysts and maxillary prognathism. Thickening of the lips and a bulbous nose have been described in many cases. Perioral linear telangiectases and cases of granulomatous cheilitis and granulomatous gingivitis are noted. Tinnitus is reported in approximately 40% of patients with this disorder. Because of the severe major organ dysfunction, most of these patients die at an early age secondary to stroke, end-stage renal failure, or cardiovascular complications.
Differential diagnosis
Because this disorder is rare, it is often misdiagnosed because of early nonspecific manifestations, including neurologic and skin disorders. These disorders can include rheumatoid arthritis, fibromyalgia, multiple sclerosis, and Meniere disease. The disorder is often discovered following more serious complications such as stroke, cardiac insufficiency, and renal failure. Because treatment with enzyme replacement therapy has been shown to have positive results, early diagnosis of Fabry disease is important. When suspected, serum assays of leukocyte alpha-galactosidase A activity are effective in men; however, genetic testing in women is recommended because of inclusive serum results.
Treatment considerations for the oral and maxillofacial surgeon
Oral surgeons should be suspicious that any patient displaying angiokeratomas of the face, cheilitis granulomatosa, or granulomatous gingivitis may have the potential for Fabry disease, especially young men. With late-stage disease and renal, cardiac, and neurologic complications, treatment must be carefully administered with close consultation with the appropriate specialists.
Williams syndrome
- •
Williams syndrome, also known as Williams-Beuren syndrome, is a rare autosomal dominant neurodevelopmental disorder affecting men and women equally.
- •
Williams syndrome was first discovered in 1961 by J.C.P. Williams, who found patients with supravalvar aortic stenosis, facial anomalies, and mental retardation.
- •
Independently in 1962, A.J. Beuren found similar phenomena in his patients but also certain dental characteristics and pulmonary artery stenosis.
Genetics
Williams syndrome and its manifestations seem to result from a 1.5-Mb deletion on the number 7 chromosome pair at 7q11.23, which includes 26 to 28 genes. This deletion significantly affects the production of elastin, which leads to supravalvar aortic stenosis and other cardiovascular disorders such as peripheral artery stenosis. The other characteristics of this disease arise because adjacent genes responsible for facial and neural development are located in the deletion. The prevalence of this mutation seems to be in the range of 1 in 7500 to 1 in 25,000 live births.
Clinical features
Williams syndrome is characterized by its elfin facial appearance, growth delays with low body weight, mental retardation, cardiovascular disorders, sleep disorders, and transient hypercalcemia, usually as infants. These patients tend to have intelligence quotients in the 50 to 60 range and are overly friendly, extremely talkative, with severe bouts of anxiety. The facial abnormalities ( Fig. 3 ) include periorbital fullness, full upper and lower lips, long philtrum, flat nasal bridge, micrognathia, and abnormal dentition ( Fig. 4 ) including hypocalcification of the enamel, short and narrow primary teeth, and crowded permanent teeth. Hyperacusis has been noted to be present in most patients. Although it is been reported that there is an increase in gingival height and width of these patients, there was no increase in loss of attachment. There have been reports in the literature of keratoconus, multiple sclerosis, Crohn’s disease, and Burkitt’s lymphoma, but these are rare.
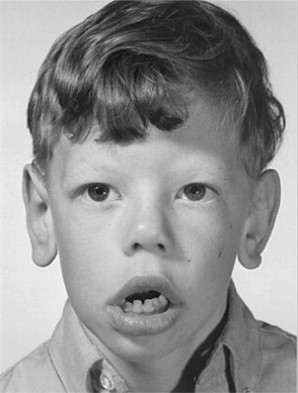
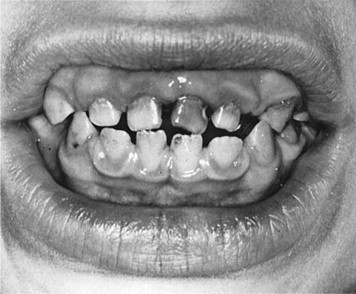
Differential diagnosis
Some of the symptoms of Williams syndrome are also found in other disorders. Patients with Noonan syndrome have similar facial appearances, including micrognathia, flat nasal bridge, and leprechaunism (an endocrine disorder that produces elfin facial features and growth retardation). Idiopathic infantile hypercalcemia not related to Williams syndrome has been reported. The definitive diagnostic test for Williams syndrome is the fluorescence in-situ hybridization test, which conclusively shows the gene deletion.
Treatment considerations for the oral and maxillofacial surgeon
Although there is no cure for Williams disease, treatment of the underlying causes becomes paramount. Management of these patients can be difficult because of their mental disorders, which often require sedation or general anesthesia for treatment. As patients age, cardiovascular concerns along with hypertension require close consultation with the appropriate medical practitioners.
Hurler syndrome
- •
Hurler syndrome is a rare autosomal recessive disease that is classified in a group of disorders called mucopolysaccharidosis. It is also known as mucopolysaccharidosis type 1-H.
- •
First described by Dr Gertrud Hurler in 1919, Hurler syndrome was discovered in 2 unrelated boys with similar characteristics including mental impairment, distortion of facial features, respiratory and cardiac disease, hepatosplenomegaly, and corneal opacities.
- •
It is the most severe form of mucopolysaccharidosis and, without treatment, leads to early death before the second decade of life.
Genetics
Hurler syndrome occurs when there is a mutation in the alpha l -iduronidase (IDUA) gene. IDUA is required for the degradation of heparan sulfate and dermatan sulfate in the lysosome. With the accumulation of these glycosaminoglycans in various organs, multiple systemic effects begin to occur over a short period of time. It seems that patients with the serious form of the disease have nonsense mutations identified on both alleles. The incidence of Hurler syndrome is about 1 in 100,000 live births.
Clinical features
Children with Hurler syndrome initially appear nearly normal at birth but, because of accumulation of glycosaminoglycans, progressive cell and tissue malfunction occurs, leading to mental retardation, hepatosplenomegaly, corneal clouding, with eventual cardiopulmonary failure. Clear cells (Hurler cells) have been identified in infants within the myocardium. Affected individuals have short long bones and short and broad digits consistent with dwarfism. Carpal tunnel syndrome and limited mobility are common. Facial deformities ( Figs. 5 and 6 ) are prevalent, including an enlarged deformed head with frontal bossing, a flattened nasal bridge, and hypertelorism. The patients have thickened patulous lips with a flattened philtrum. They have significant macroglossia that, over time, leads to an anterior open bite and, combined with a short neck, leads to sleep apnea and significant complications from anesthesia. There are dental anomalies, including short conical teeth, widely spaced dentition, and supernumerary teeth in the mandibular rami. There have been reports of increased dentigerous cyst formation in the mandible.
Differential diagnosis
At present 7 different types of mucopolysaccharidosis have been identified. Of the mucopolysaccharidosis type 1 (MPS 1) types, the phenotypes of each determines the specific syndrome. As mentioned earlier, Hurler syndrome (MPS 1-H), with the most extensive symptoms, is distinguished from Hurler-Scheie syndrome (MPS 1-H/S), with intermediate symptoms (no cognitive disturbances), versus the Scheie syndrome (MPS 1-S), with attenuated symptoms that occur later in life and progress slowly. Because early diagnosis before the age of 2 years is critical for the long-term survival of a patient with Hurler syndrome, recognition becomes paramount. Early symptoms such as rhinitis and hernia are vague, but radiographic features can aid in the diagnosis. A decreased level of serum alpha- l -iduronidase confirms the diagnosis.
Treatment considerations for the oral and maxillofacial surgeon
A patient diagnosed with Hurler syndrome before the age of 2 years has shown considerable improvement of symptoms, especially the neurologic defects, with hematopoietic stem cell transplantation. However, there are significant risks, with increased morbidity and mortality. Enzyme replacement therapy (ERT) with laronidase, which is a protein analogous to human alpha iduronidase, is an alternative therapy. However, it does not cross the blood-brain barrier and therefore shows no improvement in the neurologic deficits that the syndrome displays. For the oral and maxillofacial surgeon, these patients are extremely difficult to treat because of their neurologic disorders, skeletal deformities, dental anomalies, and anesthetic risks. Close consultation with the appropriate specialists and probable palliative treatment is indicated.
Hunter syndrome
- •
Hunter syndrome, also known as mucopolysaccharidosis II, is a rare X-linked lysosomal storage disorder.
- •
Hunter syndrome was first described by Dr Charles Hunter in 1917, who described 2 brothers with significant skeletal deformities, including facial deformities with hearing deficiencies.
- •
The syndrome causes a deficiency in the lysosomal enzyme iduronate-2-sulphatase, which is responsible for cleaving sulfate from the glycol aminoglycosides dermatan sulfate and heparin sulfate. As a result, gradual accumulation of these glycosaminoglycans results in multiple organ and systemic disorders.
Genetics
The incidence of mucopolysaccharidosis II (MPS II) ranges from 1 in 110,000 to 1 in 135,500 live births. Because it is an X-linked disease, most patients are male. There are some female cases reported, which require either 2 pathologic alleles or a normal inactivated allele. However, the female cases tend to have attenuated symptoms. The gene that produces iduronate-2-sulphatase is located on chromosome Xq28 with the exonic point mutations G374sp comprising half of the mutations. Significant variability in the mutations is evident. It is thought that this variance explains much of the difference between phenotypes and also the difficulty in treating patients with MPS II.
Clinical features
Similar to other patients with mucopolysaccharidosis, patients with MPS II tend to appear normal at birth. Frequent upper respiratory infections, umbilical hernias, and coarse facial features are some of the early signs and symptoms. As the increase in glycosaminoglycosides accumulate in organs, hepatosplenomegaly, skeletal and joint involvement with decreased mobility, clubbed fingers, cardiac involvement, and obstructive airway disease become prevalent. Facial features ( Fig. 7 ) include frontal bossing, a wide nasal bridge, a large skull, macroglossia often leading to an anterior open bite, and mandibular immobility. Non-tender, irregularly shaped papules may be present on the skin of the upper back ( Fig. 8 ). Dental abnormalities have been reported, including widely spaced teeth, enamel defects, carious teeth, dentigerous cysts, and abscesses. In the most severe cases of Hunter disease, central nervous system manifestations begin between the ages of 2 and 4 years and are often expressed as severe mental retardation. In these situations, death is usually expected before the third decade of life and usually as a result of cardiac or obstructive respiratory failure. In the milder forms, patients may have normal intellect and near-normal stature with an increase in life expectancy.
Differential diagnosis
Because there are significant differences in the expression of this disorder, it can be difficult to distinguish this from other forms of mucopolysaccharidosis. In the severe expression of MPS II, there are many similar characteristics to Hurler syndrome, except without the corneal clouding. Other lysosomal storage disorders that share similar features include MPS VI (Maroteux-Lamy syndrome) and MPS VII (Sly syndrome). A preliminary test to confirm mucopolysaccharidosis is an increase in urinary glycosaminoglycans. Although this is not specific to Hunter syndrome, further blood testing for deficiency of iduronate-2-sulfatase in leukocytes, serum, or plasma will secure the diagnosis. A mnemonic screening tool has been developed and has correctly identified 95% of patients with no family history of Hunter syndrome.
Treatment considerations for the oral and maxillofacial surgeon
With many mucopolysaccharidosis disorders, early diagnosis is critical because estrogen replacement therapy (ERT) with idursulfase has been shown to be effective in delaying and improving many of the symptoms associated with this disorder. However, this drug does not seem to cross the blood-brain barrier and therefore has little effect on those patients with severe central nervous system involvement. Unlike Hurler syndrome (MPS 1-H), hematopoietic stem cell transplantation does not seem to have any significant effect on neurologic improvement. The oral and maxillofacial surgeon must be cognizant that each individual with Hunter syndrome may present with a variety of symptoms. Anesthetic management and risks associated with individual patients become paramount.
Heerfordt syndrome
- •
Heerfordt syndrome is a rare manifestation of systemic sarcoidosis that includes fever, parotid swelling, uveitis, and facial nerve paralysis.
- •
It was described by Danish ophthalmologist Christian Heerfordt, who reported these traits without the connection to sarcoidosis. That direct link was reported in 1937 by Dr Jan Waldenstrom.
- •
The histology is classic for multiple noncaseating granulomas found in the parotid or salivary glands and lacrimal glands.
- •
Treatment of Heerfordt syndrome with long-term corticosteroids seems to have a good prognosis.
Genetics
Sarcoidosis and therefore Heerfordt syndrome seems to be more prevalent in women than in men, and in black Americans versus white with 3 times the rate (36 per 100,000 vs 11 per 100,000) and also with more chronic and fatal disease. Most patients show clinical signs between the ages of 20 and 40 years. Numbers vary around the world but it seems that the frequency of Heerfordt disease is about 0.3% of patients with sarcoidosis. Because sarcoidosis seems to have a genetic and environmental component, it is thought that Heerfordt syndrome has similar findings. The human leucocyte antigen (HLA) gene has been implicated in both sarcoidosis and in Heerfordt syndrome but specifically the HLA-DRB1*04 allele seems protective for sarcoidosis, but increases the risk for ocular manifestations. Despite significant research the exact cause has remained elusive and further genetic and immunologic studies are required.
Clinical features
In patients with sarcoidosis, usually fever, followed by uveitis, parotid gland swelling, and then facial nerve palsy are classic for Heerfordt syndrome ( Figs. 9 and 10 ). The parotid glands usually enlarge bilaterally and are firm to the touch. Bilateral lacrimal gland enlargement is often seen with either anterior or posterior uveitis, accompanied by blurred vision. Although the facial nerve palsy is usually unilateral, bilateral deficits have been reported. Neurosensory deficits of other cranial nerves, including the palate, have been reported.
Differential diagnosis
The differential diagnosis for each of the components for Heerfordt syndrome can be extensive. For example, bilateral parotid swelling can be attributed to Sjögren syndrome, tuberculosis, mumps, sialadenitis, and Mikulicz disease; however, none of these have uveitis. The definitive diagnosis is made by histologic confirmation of sarcoidosis, which presents as noncaseating granulomas. In cases in which tissue biopsy is difficult, increased angiotensin converting enzyme levels in combination with a positive 67 gallium scintigraphy have been shown to have 99% accuracy.
Treatment considerations for the oral and maxillofacial surgeon
Patients presenting with any of the symptoms of Heerfordt syndrome should immediately be referred for the evaluation of sarcoidosis. The treatment of this disorder is usually corticosteroids and occasionally azathioprine. Long-term prednisone is often required. The long-term prognosis for these patients is usually good, but a small number (less than 5%) die from sarcoidosis, usually as a result of pulmonary fibrosis with respiratory failure.
Sjögren syndrome
- •
Sjögren syndrome is a chronic autoimmune inflammatory disease that primarily affects the exocrine glands. It was first described by Swedish ophthalmologist Henrik Sjögren in 1933.
- •
The syndrome can manifest as a primary disorder usually within the lacrimal and salivary glands or have systemic manifestations (most commonly rheumatoid arthritis or systemic lupus erythematosus), and is defined therefore as secondary Sjögren.
- •
Although the exact cause is unknown, it seems that genetic, environmental, and hormonal factors play a significant role.
- •
Patients with this disorder are at high risk for the development of non-Hodgkin lymphoma.
- •
Treatment of this disorder is mainly symptomatic, although immunologic therapies have shown some promise.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


