Papillon-Lefevre syndrome
- •
Periodontal disease associated with Papillon-Lefevre syndrome (PLS) affects the primary and permanent dentition.
- •
Keratosis of the palms and soles and the dorsal surfaces and other skin sites are characteristic components of the syndrome.
- •
Cathepsin C gene mutations result in a gene product that does not have functional cathepsin C (CTSC) activity.
- •
PLS and Haim-Munk syndrome (HMS) are allelic variants of cathepsin C gene mutations.
Genetics
Papillon-Lefevre syndrome (PLS) is an autosomal-recessive disorder caused by mutations on the cathepsin C gene. The cathepsin C (CTSC) gene mutations have been mapped to 11q14.1-14.3. Heterozygous carriers of the mutation do not have clinical manifestations of the disease. CTSC is a lysosomal protease and functions as an activator of neutrophil serine proteases. Defective CTSC function likely impairs microbial degradation, cytokine pathways, neutrophil recruitment, and macrophage dysfunction. Also, impaired natural killer cell cytotoxicity is noted in PLS. Haim-Munk syndrome and perhaps aggressive prepubertal periodontitis are similar to PLS in that they too demonstrate inactivation of CTSC. A late onset variant of PLS without alteration of the CTSC gene has been described, but this clinical situation is likely due to another genetic cause.
Clinical features
The syndrome was first described by Papillon and Lefevre in 1924. The clinical findings associated with PLS include palmoplantar erythema and hyperkeratosis, and severe periodontitis that affects both the primary and the permanent dentitions ( Figs. 1 and 2 ). Haim-Munk syndrome, described in 1965, has in addition to the findings that characterize PLS, atrophic changes to the nails, and finger findings of acro-osteolysis and clawlike volar curves noted radiographically. Susceptibility to infection has been described in these hereditary forms of palmoplantar keratoses. Tooth loss is preceded by gingival inflammation and subsequent periodontal bone loss and tooth mobility; Aggregatibacter actinomycetemcomitans (AA) is an identified pathogen in PLS. Tooth loss follows the pattern of eruption, incisors lost first, and then the more posterior dentition. Both primary and permanent dentition are affected. The permanent dentition is often lost in the teenage or early adult years. Upon tooth loss, the alveolar mucosa is normal in appearance. Gorlin reported calcified falx and choroid plexus in PLS.
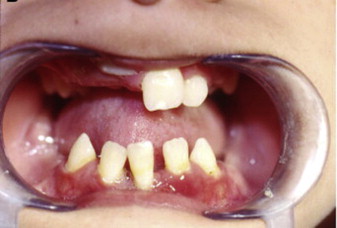
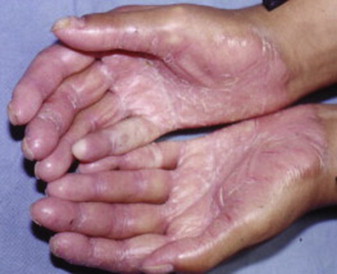
Differential diagnosis
All forms of aggressive periodontitis should be included in the differential. These forms of aggressive periodontitis include syndromes associated with decreased number of neutrophils, including the various types of severe congenital neutropenia syndromes, cyclic neutropenia, bone marrow failure syndromes, as well as syndromes with abnormal neutrophil function, such as Chediak-Higashi syndrome, and leukocyte adhesion deficiency types 1 and 2. In addition, syndromes of metabolic or structural defects, such as Kindler syndrome, Ehlers-Danlos syndrome type IV and VIII, hypophasphatasia, and hypotrichosis-osteolysis-periodontitis-palmoplantar keratoderma syndrome should also be included in the differential.
Treatment considerations
Careful attention to oral hygiene and periodontal care are essential; however, disease is progressive even with conventional approaches to periodontal disease. It has been recommended that compromised primary teeth should be extracted 6 months before eruption of the permanent dentition. Ishikawa reported suspect bacterial pathogens, including AA and a benefit of extraction of the primary dentition, but the permanent dentition status was followed only for several years. Antimicrobial therapy has been tried, including amoxicillin and metronidazole regimens for periodontal disease as well as treatment with retinoids for dermatologic manifestations. Retinoids have not consistently proved effective for periodontal disease. Various studies of implant placement in the PLS patient have been described, yet too few to form solid recommendations.
Hereditary gingival fibromatosis type 1
- •
Hereditary gingival fibromatosis type 1 (HGF1) is a benign progressive fibrous gingival enlargement.
- •
HGF1 is the result of a mutation in the SOS1 gene on chromosome 2p21.
- •
HGF1 severity is variable among affected individuals; severe forms can impede tooth eruption.
- •
Gingival fibromatosis is a feature of many syndromes.
- •
Gingival hyperplasia related to medications (anticonvulsants, calcium channel blockers, and cyclosporine) can clinically simulate HGF1.
- •
HGF1 treatment consists of gingivectomy; timing of the procedure is controversial.
Genetics
Hereditary gingival fibromatosis 1 (GINGF1) is an autosomal-dominant form of gingival overgrowth caused by a heterozygous frameshift mutation in the SOS1 gene on chromosome 2p21. Additional forms of HGF (GINGF2, GINGF3, and GINGF4) have been mapped to other chromosome loci. A less common autosomal-recessive form and sporadic cases of HGF are recognized. Mutation on the SOS1 gene has also been reported in Noonan syndrome. The exact mechanism by which the SOS1 gene mutation results in the gingival overgrowth is not currently known.
Clinical features
HGF1 is characterized by slowly progressive fibrous overgrowth of the gingival tissues of the maxilla and mandible ( Fig. 3 ). Typically, the overgrowth is nonhemorrhagic and the affected tissues are firm and of normal color, but secondary inflammatory changes can occur. The condition manifests at the time of eruption of the primary or permanent dentition, and the degree of gingival overgrowth can be variable. Delayed tooth eruption may occur. This phenomenon is referred to as variable expressivity. Thus, members of an affected family may demonstrate varying degrees of severity of the gingival overgrowth. Severe cases may completely cover the dentition. A kindred with HGF and associated hypertrichosis has been described. The clinical presentation of HGF1 may be indistinguishable from syndromes that may have gingival overgrowth as a component of the disease and gingival overgrowth secondary to particular medications known to cause gingival hyperplasia. The histopathology of HGF1 is characterized by abundant collagen with interspersed spindle-shaped fibroblasts, typically without an inflammatory component, but a chronic inflammatory component consisting of plasma cells and lymphocytes may be a secondary finding. The surface stratified squamous epithelium often exhibits elongation of the rete pegs into the underlying collagenous stroma. The histopathology is nonspecific and similar to the pathologic abnormality noted in syndrome-related and medication-related gingival overgrowths.
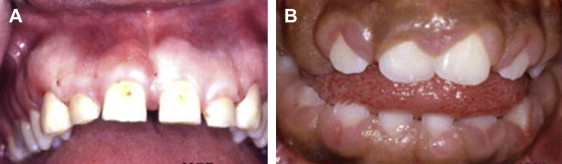
Differential diagnosis
A comprehensive review of syndromes that may have gingival fibromatosis has been published by Hart and colleagues. These syndromes include but are not limited to Jones syndrome (gingival fibromatosis with progressive deafness), gingival fibromatosis with hypertrichosis, gingival fibromatosis with distinctive facies, Ramon syndrome, Zimmerman-Laband syndrome, juvenile hyaline fibromatosis, Cross syndrome, and Rutherfurd syndrome. Katz and colleagues reported a case of gingival fibromatosis and associated supernumerary tooth, chest deformity, auricular cartilage deformation, joint laxity, and undescended testes. Wynne and colleagues reported HGF with associated hearing loss and supernumerary teeth. Medication-related gingival hyperplasia also enters into the differential diagnosis. Anticonvulsant medications, calcium channel blockers, and the immunosuppressive medication cyclosporine, as well as oral contraceptives, can result in gingival overgrowth. The diagnosis of HGF requires an adequate history and clinical examination with the exclusion of the aforementioned related conditions.
Treatment considerations
Gingivectomy is the recommended treatment. It has been suggested that this is best accomplished after the permanent teeth have erupted. However, severe cases of HGF may require gingivectomy to allow the dentition to erupt when the gingival overgrowth is thought to be responsible for lack of anticipated tooth eruption. Repeated debulking of the affected gingival tissues may be necessary.
Hereditary gingival fibromatosis type 1
- •
Hereditary gingival fibromatosis type 1 (HGF1) is a benign progressive fibrous gingival enlargement.
- •
HGF1 is the result of a mutation in the SOS1 gene on chromosome 2p21.
- •
HGF1 severity is variable among affected individuals; severe forms can impede tooth eruption.
- •
Gingival fibromatosis is a feature of many syndromes.
- •
Gingival hyperplasia related to medications (anticonvulsants, calcium channel blockers, and cyclosporine) can clinically simulate HGF1.
- •
HGF1 treatment consists of gingivectomy; timing of the procedure is controversial.
Genetics
Hereditary gingival fibromatosis 1 (GINGF1) is an autosomal-dominant form of gingival overgrowth caused by a heterozygous frameshift mutation in the SOS1 gene on chromosome 2p21. Additional forms of HGF (GINGF2, GINGF3, and GINGF4) have been mapped to other chromosome loci. A less common autosomal-recessive form and sporadic cases of HGF are recognized. Mutation on the SOS1 gene has also been reported in Noonan syndrome. The exact mechanism by which the SOS1 gene mutation results in the gingival overgrowth is not currently known.
Clinical features
HGF1 is characterized by slowly progressive fibrous overgrowth of the gingival tissues of the maxilla and mandible ( Fig. 3 ). Typically, the overgrowth is nonhemorrhagic and the affected tissues are firm and of normal color, but secondary inflammatory changes can occur. The condition manifests at the time of eruption of the primary or permanent dentition, and the degree of gingival overgrowth can be variable. Delayed tooth eruption may occur. This phenomenon is referred to as variable expressivity. Thus, members of an affected family may demonstrate varying degrees of severity of the gingival overgrowth. Severe cases may completely cover the dentition. A kindred with HGF and associated hypertrichosis has been described. The clinical presentation of HGF1 may be indistinguishable from syndromes that may have gingival overgrowth as a component of the disease and gingival overgrowth secondary to particular medications known to cause gingival hyperplasia. The histopathology of HGF1 is characterized by abundant collagen with interspersed spindle-shaped fibroblasts, typically without an inflammatory component, but a chronic inflammatory component consisting of plasma cells and lymphocytes may be a secondary finding. The surface stratified squamous epithelium often exhibits elongation of the rete pegs into the underlying collagenous stroma. The histopathology is nonspecific and similar to the pathologic abnormality noted in syndrome-related and medication-related gingival overgrowths.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


