▪
BENZODIAZEPINES
DISCOVERY AND DEVELOPMENT
During the summer of 1949, neurochemistry researcher Eugene Roberts was working in a laboratory in Bar Harbor, Maine, studying the amino acid content of neuroblastomas in mice. Using paper chromatography, Roberts analyzed brain extracts of mice with neuroblastoma to compare them with mice that were tumor free. He discovered a chemical that was present in the tumor-free mice and was absent when neuroblastoma was present. This chemical was soon afterwards identified as γ-amino butyric acid (GABA), but its role was unclear at the time. It was not until 1957 that GABA was identified as an important inhibitory neurotransmitter of the central nervous system (CNS).
The 1950s also represented an earnest search for sedative-hypnotics that could work as effectively as the barbiturates and phenothiazines but with fewer side effects and lower abuse potential. Dr. Leo Sternbach, a scientist at Hoffman-LaRoche, was working with a class of compounds that held promise as barbiturate replacements, but found only one compound in this group that had any chemical activity. The compound was shelved and forgotten for several years, and, while cleaning his laboratory, Sternbach came across the compound and decided to test it further. This compound was chlordiazepoxide, a benzodiazepine (BZD)—it was found to have sedative and muscle relaxant properties and later to also possess anticonvulsant and amnestic effects. It was approved by the FDA in 1960 and marketed by Hoffman-LaRoche as Librium, the prototype of a drug group that would dramatically change the landscape of anesthesia practice.
The connection between the BZD and GABA was not fully elucidated until the 1970s when GABA receptors were identified in the CNSs of mammals. This led to the development of additional BZD agonists and the search for antagonists. The first true antagonist, flumazenil, was synthesized in 1979.
STRUCTURE AND ACTIVITY OF BZD
All BZD are characterized by having one benzene ring fused to a seven-membered diazepine ring. They act in two locations: at central receptors on neuronal membrane and peripherally on receptors on mitochondria. The pharmacologic actions of the BZD are closely related to the inhibitory neurotransmitter GABA. GABA exerts its inhibitory effect by opening the chloride ion channel on neuronal membranes, thus hyperpolarizing the cell membrane and making it less likely to depolarize. BZD bind to benzodiazepine receptors presynaptically and postsynaptically, which facilitates the binding of GABA and potentiates its activity, causing CNS inhibition. In the absence of GABA, the BZD have no innate pharmacologic effects.
The GABA-BZD receptor complex has been studied extensively and, although the precise three-dimensional configuration has not been mapped, there is general agreement that it is a macromolecular complex containing at least 16 different types of subunits. These subunits include GABA A receptors, a BZD receptor, a barbiturate receptor, and a chloride ionophore (channel). A BZD receptor must be linked to GABA A receptors, but not all GABA A receptors are paired with BZD receptors. It is still unknown why there exists a BZD receptor because to date there have been no endogenous BZD-like chemicals identified. All BZD, both agonists and antagonists, bind to the receptors reversibly and competitively.
Other drugs have been found to bind to the GABA-BZD receptor complex, although their relationships to GABA activity do not appear to be as specific as the BZD. These include barbiturates, propofol, steroids, etomidate, and inhalation anesthetic agents. Other anesthetics, such as ketamine and nitrous oxide, appear to have no activity at the GABA-BZD receptor complex.
CLINICAL EFFECTS
The pharmacodynamic effects of BZD have been well characterized and include anxiolysis, sedation-hypnosis, muscle relaxation, amnesia, and anticonvulsant activity. Each BZD has a differing affinity for the receptor, causing differences in potency and pharmacokinetics between drugs. Nevertheless, regardless of the BZD administered, the clinical effects are similar between drugs and are closely related to receptor saturation. At present the clinical effects are linked and one cannot administer a BZD to achieve one therapeutic effect (e.g., anxiolysis) without risking amnesia or sedation. However, there are ongoing research efforts to identify and isolate compounds that can selectively create anxiolysis alone without creating amnesia, sedation, or muscle relaxation. As more is learned about the BZD receptor subunits, it may provide future clinicians with classes of drugs that can selectively provide anxiolysis with no other effects.
Anxiolysis is the first therapeutic effect achieved when titrating the BZD. There are few studies that support any particular BZD as being most effective in creating anxiolysis. The level of anxiolysis is dose related. Older textbooks instructed clinicians to titrate a BZD until Verrill’s sign was noted (i.e., relaxation causing ptosis of the eyelids to the point that the lower border of eyelid bisected the pupil). However, Verrill’s sign is a sedative—not an anxiolytic—end point, and it is possible that some patients will have benefited sufficiently from anxiolytic doses of the BZD that there would be no need to administer additional drug toward a sedative end point. The inherent benefit of titrating to an anxiolytic end point would be to minimize the total drug dose and therefore decrease the time for recovery. The liability of this practice, however, is that amnesia to the surgery would occur less predictably.
The sedative-hypnotic effects of the BZD are linearly related to dose and receptor saturation. Anxiolysis is produced when less than 20% of receptors are occupied: Sedation occurs when there is 30% to 50% occupancy and unconsciousness when greater than 60% of receptors are occupied.
Anterograde amnesia is a well-documented effect of the BZD and is dependent on dose and route of administration. For example, diazepam 10 mg given orally can provide anxiolysis and sedation, but only 40% of patients will have some degree of amnesia.
For greater than 70% of patients to be amnestic, oral doses of 20 mg or more would have to be administered. Intravenously, doses of 5 mg and 10 mg of diazepam can produce amnesia in 50% and 90% of patients, respectively. The incidence is similar with midazolam, with complete amnesia in 67% of patients who received 5 mg midazolam intravenously.
Patients often appreciate being amnestic to an oral surgical procedure, and the degree of amnesia roughly follows in a linear fashion the depth of sedation. However, despite an adequate depth of sedation, it is possible there still may be recall, and there are no reliable predictors to tell us which patients may remember portions of the surgery. Therefore unless absolute general anesthesia is planned throughout the entire surgery, it would be wise to inform patients that awareness during the anesthetic may occur and the patient may retain some memory of the surgery.
The muscle relaxant effect of the BZD occurs both centrally and at the spinal cord. Glycine is the major inhibitory neurotransmitter in the brainstem and spinal cord, and BZD mimic glycine thus causing decreased muscle tone. The actions of the BZD are not related to the motor end plate or neuromuscular transmission.
The BZD depress the central respiratory drive and CO 2 response curve. When BZD are used alone and in typical doses to achieve conscious sedation, the respiratory depressant effects rarely cause clinically significant hypoventilation or apnea. However, when used with other agents (e.g., opioids) or when used in elderly or medically compromised patients, the respiratory depressant effects can be significant and long lasting. It was in elderly patients undergoing endoscopic procedures that the respiratory depressant effects of midazolam were first noted to be potentially dangerous, primarily because of respiratory arrest. For diazepam there may be a 50% to 65% decrease in minute ventilation, and for midazolam it can be anywhere from a 25% to 65% decrease that can last for hours.
When used alone in healthy patients, BZD cause very slight changes in heart rate, blood pressure, left ventricular end diastolic pressure, and systemic vascular resistance. This cardiovascular stability is thought to result from the maintenance of normal baroreceptor function. Diazepam has a beneficial effect on coronary blood flow, increasing it 22% in healthy patients and 73% in patients with coronary disease. Midazolam neither increases nor decreases coronary blood flow.
Whereas the effects of BZD in younger patients may not become clinically significant, the elderly are much more sensitive to these effects and may, in fact, show significant cardiovascular changes. Even with modest doses and with minor surgical procedures, a significant number of patients can experience hypotension after administration of midazolam.
When the BZD are combined with other drugs (e.g., opioids), there may be significant changes in mean arterial pressure, systemic vascular resistance, cardiac index, and stroke volume.
PHARMACOKINETICS AND METABOLISM
Plasma concentrations of BZD follow a linear to slightly sigmoid dose-response curve. All are highly bound to plasma proteins, with slight differences between drugs ( Table 4-1 ). Metabolism occurs in the liver through glucuronide conjugation and hepatic microsomal enzymes. Because liver function can diminish with age and with disease, metabolism can be expected to occur more slowly in the elderly and in patients with cirrhosis or hepatitis. Concomitant administration of certain drugs can also increase elimination time; this includes erythromycin, oral contraceptives, and calcium channel blockers. All BZD are eliminated through the kidneys.
| Elimination ½ Life (hr) | % Protein Bound | |
|---|---|---|
| Midazolam | 1–5 | 95 |
| Diazepam | 20–40 | 97 |
| Lorazepam | 10–20 | 88-92 |
| Triazolam | 1–5 | 85-90 |
| Alprazolam | 9–15 | 70 |
| Flumazenil | 0.7–1.3 | 54-64 |
The BZD have among the widest toxic-therapeutic ratios of any agent used for sedation and anesthesia, making them a safe drug class. There have been reported cases of ingestion of 2000 mg diazepam orally by an adult and 300 mg by a 2-year-old, with complete recovery in both cases.
The only true contraindication to the use of any BZD is allergy to the drug and acute narrow-angle glaucoma.
Diazepam
In 1965 the introduction of diazepam to dental practice greatly improved the dentist’s ability to provide conscious sedation. Compared with the multidrug regimens that were commonly used, diazepam represented a drug that could be very effective as the sole sedative and anxiolytic agent.
The pH of diazepam ranges from 6.2 to 6.9 and has an elimination half-life of more than 40 hours.
Because it is not water soluble, a number of different solvents have been used over the years to create a preparation for parenteral use. The preparation approved by the FDA included the solvent propylene glycol, whose irritating properties cause the risk of phlebitis and venous thrombosis. To minimize this risk, the manufacturer’s package insert cautions that the drug must be administered slowly—no more than 1 mL (5 mg) per minute—and it should not be injected into small veins, such as the dorsum of the hand or the wrist. The injectable solution also contains 10% ethanol, 5% sodium benzoate (a buffer), and 1.5% benzyl alcohol (a preservative). For several years, a preparation of diazepam became available that used a solvent containing the same lipid emulsion as propofol (Dizac), but this product was discontinued in 2000.
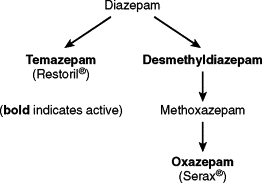
Diazepam is metabolized into several other BZD, some of which have sedative properties ( Fig. 4-1 ). The long half-life of the drug and the presence of active metabolites make this drug less than ideal for use in elderly patients or those who are medically compromised.
Midazolam
Midazolam was synthesized in 1975 and introduced for clinical use in 1986, offering several notable advantages over diazepam: It had a much shorter half-life, did not have active metabolites, and was water soluble. This provided shorter recovery time for patients and avoided the injection of solvents that were irritating to veins. Midazolam’s water solubility derives from an imidazole ring that can selectively open and close, depending on ambient pH. At an acidic pH of less than 4.0, the ring is open, conferring hydrophilicity to the molecule and thus making it water soluble. Hence midazolam is packaged in vials that have an adjusted pH of 2.9 to 3.7. Once the solution is injected and is exposed to plasma pH of 7.4, the ring closes and the molecule becomes lipophilic and is able to bond to receptors on the neural membrane. The ring closure is not instantaneous, and complete closure may not occur for 5 to 10 minutes after injection. Because the clinical effects cannot occur until the ring has closed, peak drug effects may not be seen immediately after injection.
Its water solubility has permitted midazolam liquid to be administered intramuscularly, nasally, rectally, and orally, routes that were not available for use with diazepam. Oral administration of midazolam was first investigated in 1988, and a number of clinical studies confirmed its effectiveness in children. In 1997 the FDA approved the use of midazolam in children, and it became available in an oral syrup form in 1998.
Triazolam (Halcion)
This BZD was originally marketed in the Netherlands in 1977 as a hypnotic and suspended from use after allegedly causing “raving madness” in some patients, but a thorough investigation failed to provide sufficient evidence to keep the drug off the market. A similar concern was raised in the United States, but it was never removed from the market, although the manufacturer discontinued manufacturing the highest dose (0.5 mg) tablet.
It has a rapid onset of action (1 hour), a short elimination half-life, and is eight times more effective than diazepam as a hypnotic. Because of its favorable pharmacokinetics, it has remained popular as an oral premedicant before outpatient surgical procedures. Most recently there has been increased attention focused on sublingual administration of triazolam. Because the first-pass effect through the liver is eliminated compared with traditional oral administration, there is a 28% increase in bioavailability of the drug when given sublingually compared with orally, causing greater anxiolysis and amnesia.
Lorazepam (Ativan)
This drug, frequently used in intensive care units for prolonged sedation, does not have a favorable profile for ambulatory anesthesia. It has a short distribution half-life and a long elimination half-life. As a result of its lower lipid solubility, its effects do not reach a peak until 30 to 60 minutes after the initial IV dose. It is more effective at producing amnesia than diazepam, and the amnestic effects may last many hours after administration.
Flumazenil
At the time the most popular BZD, diazepam and midazolam, were introduced for clinical use, there were no specific antagonists available. Nonspecific antagonists, physostigmine or aminophylline, were used to reverse the effects of BZD. The “reversal” effects were actually generalized CNS excitation that frequently would overshadow some of the sedative effects of the BZD. However, this was not true reversal because no specific action occurred at the BZD receptor. Flumazenil, the first true antagonist, was synthesized in 1981 and introduced into clinical practice in 1992. Flumazenil is an imidazobenzodiazepine that interacts with GABA-BZD complex and competitively displaces the BZD. It is not protein bound, and its half-life is roughly 1 hour, making resedation a risk when longer acting BZD or high doses have been administered. Flumazenil reverses all BZD effects, but does not antagonize the effects of other drugs that affect the GABA receptors, such as ethanol, barbiturates, opioids, or ketamine.
Flumazenil is a very safe drug, with extremely high doses reported to be given with generally no adverse effects. The few adverse effects that have been reported, including anxiety, crying, tachycardia, and nausea and vomiting, are likely related to the sudden reversal of the therapeutic effects of the BZD.
▪
OPIATES AND OPIOIDS
DISCOVERY AND DEVELOPMENT
Archaeologic evidence and written records confirm that the pharmacologic effects of the juice from the opium poppy, papaver sominferum , were known to prehistoric man and early ancient civilizations. Over centuries of time, opium was used as an analgesic, antidepressant, antidiarrheal, and cough suppressant. The drug was ingested orally as a tea or mixed with alcoholic beverages, and it was smoked.
In 1805 morphine was isolated from opium in Germany. It and its methylated derivative codeine are still produced today directly from commercially grown opium. In the mid-1800s when the syringe and needle became available, morphine was the first drug injected using the newly designed syringe. Hence by the time of the Civil War, it was possible to treat soldiers injured in combat by injecting morphine.
The first semisynthetic opioids were heroin and hydromorphone (Dilaudid), produced by making simple chemical changes to morphine’s piperidine ring, the portion that confers its pharmacologic activity. The next breakthrough was the synthesis of meperidine, discovered during the search for an atropine-like drug. It was serendipitously learned that meperidine, in addition to antimuscarinic effects, also possessed opiate-like effects even though its chemical structure was different. Later, the phenylpiperidine structure of meperidine was used as the basis for creating a whole series of synthetic opioids beginning with fentanyl, and later alfentanil, sufentanil, and remifentanil. The only ones in the fentanyl series that have not been used for humans are carfentanil and lofentanil, whose potency is so great that they are generally considered to be useful only for veterinary management of large animals.
A class of agonist-antagonists was also developed, beginning with pentazocine (Talwin) that was introduced in 1967. The major goal of developing this class of drugs was to provide dose-dependent analgesia while limiting the adverse effects (e.g., respiratory depression). Later other agonist-antagonists were developed, including butorphanol (Stadol), nalbuphine (Nubain), buprenorphine (Buprenex), and dezocine (Dalgan).
STRUCTURE AND ACTIVITY
From the 1950s until the early 1970s, the idea of an opioid receptor had been postulated but unproven. In 1973 the first articles appeared demonstrating the presence of opiate receptors, followed by studies that demonstrated the presence of endogenous opiates. Later a number of different opiate receptor subtypes were characterized. There are three major receptors (mu, delta, kappa) with as many as eight subtypes.
The agonist-antagonists possess partial agonist activity at μ and other receptors in addition to competitive antagonist activity. This has been shown to create a ceiling effect that limits respiratory depression regardless of dose administered. It also limits the possibility of physical dependence.
The antagonists competitively bind at opioid receptors, displacing any agonists present and thus reversing the effects of the agonist.
CLINICAL EFFECTS
The primary desirable effect of opioids is analgesia. Analgesia can be created at the spinal level through activation of presynaptic opioid receptors. This leads to decreased levels of neurotransmitters and thus decreased transmission of pain-induced action potentials. Supraspinal analgesia occurs by activation of postsynaptic opioid receptors in the brainstem and midbrain, leading to hyperpolarization and thus neuronal inhibition. There may also be analgesia created through peripheral mechanisms, as evidenced by reduction of pain when morphine is injected directly into joints postoperatively.
When used as an adjuvant drug for ambulatory anesthesia, the desired effects of opioids and opiates are not necessarily analgesia but rather sedation and euphoria, both of which supplement and augment the actions of other sedative agents.
The respiratory depressant effects of the opioids are caused by a dose-dependent depression of the brainstem response to increased CO 2 and decreased O 2 . The respiratory pattern changes as well, characterized by a low respiratory rate with a large tidal volume. Respiratory depression is accentuated by a variety of factors, including increasing age, concomitant administration of other CNS depressants, circadian rhythms, and sleep deprivation.
Opioids are known to cause histamine release. There is also a central decrease in sympathetic tone, leading to a tendency toward vagal-dominated bradycardia. As a result of these two effects, direct and indirect vasodilation occurs.
Opioids can rapidly cross the placental barrier and are found at significant concentrations in the breast milk of nursing mothers. After opioid administration has ceased, it can take up to 96 hours for these concentrations to clear, making it necessary for nursing mothers to feed their infants using stored milk and to express and discard breast milk produced during this period of time.
Opioids have been associated with chest wall rigidity that usually occurs only after loss of consciousness and is associated with high doses and rapid administration. That said, there are cases reported where it occurred in an awake patient who received a small dose. The mechanism is still not completely understood. Some have concluded that it results solely from glottic closure. Others have opined that it relates to the effects of opioids on specific sites in the CNS, such as the nucleus raphe pontis in the reticular formation of the brainstem. It is not caused by direct stimulation of skeletal muscle because it can be prevented by pretreatment with muscle relaxants. Of all the opioids, fentanyl is the one most prominently implicated in causing chest wall rigidity.
Meperidine
Meperidine, the first totally synthetic opioid, is also known by its non-U.S. name “pethidine.” Its duration of action is anywhere from 3 to 5 hours.
The atropine-like effects of meperidine differ from those of other opioids: tachycardia, decreased myocardial contractility, and mydriasis. One of the metabolites, normeperidine, is long acting and pharmacologically active and can cause toxic effects in the CNS that can result in increased EEG activity, myoclonus, and seizures. These effects are not reversible by naloxone.
Of all the opioids, meperidine is the one most likely to cause histamine release. In one study patients showed as high as a 200-fold increase in serum histamine levels several minutes after its administration, a far greater amount than either morphine, fentanyl, or sufentanil. In addition to the potential to create symptoms similar to anaphylaxis, this histamine release may also be responsible for vasodilation that can lead to clinically significant hypotension.
Fentanyl
Fentanyl is 60 to 80 times more potent than morphine, and there is a 2 to 3 times greater affinity for fentanyl at the opiate receptor compared with morphine. After injection fentanyl is characterized by a rapid onset of action owing to its ease of crossing the blood-brain barrier. It is rapidly eliminated, with 99% of a single dose cleared from the plasma within 60 minutes.
Remifentanil
Remifentanil, the most recently introduced opioid, possesses unique properties by being μ-selective. It also has an ester linkage and is thus metabolized by tissue and plasma esterases. This imparts an extremely short half-life and limits the accumulation of the drug in the tissues. This pharmacokinetic profile makes it ideal for continuous infusion because a steady-state level can be reached very rapidly, and after discontinuing the infusion, the level of detectable drug in the serum drops very quickly.
Naloxone
This short-acting antagonist can be used to quickly reverse the effects of opioids. Its short duration of action, however, implies there could be recurrence of the agonist effects when a long-acting opioid was used. Naloxone is associated with nausea and vomiting, the incidence of which increases with dose and rapidity of administration. Sudden reversal of an opioid with naloxone has also been associated with rebound sympathetic stimulation that can cause dysrhythmias, hypertension, myocardial infarction, stroke, and pulmonary edema.
▪
OPIATES AND OPIOIDS
DISCOVERY AND DEVELOPMENT
Archaeologic evidence and written records confirm that the pharmacologic effects of the juice from the opium poppy, papaver sominferum , were known to prehistoric man and early ancient civilizations. Over centuries of time, opium was used as an analgesic, antidepressant, antidiarrheal, and cough suppressant. The drug was ingested orally as a tea or mixed with alcoholic beverages, and it was smoked.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


