3.3 Human Papillomavirus in OSCC
Human papillomavirus (HPV ) is strongly correlated with cervical carcinoma. HPV DNA has been identified in approximately 24 % of OSCC, with HPV-16 and HPV-18, the most common types, accounting for almost 70 % and 8 % of HPV-positive OSCC, respectively [20–22]. High-risk oncogenic HPV-16 and HPV-18 contain two oncogenes, E6 and E7, which induce cell transformation and cause cell cycle dysregulation [11]. E7 induces the proteolytic degradation of phosphorylated retinoblastoma (pRb), and E6 inhibits p53 activation by facilitating its ubiquitin-mediated proteolysis [11, 23]. Typical HPV-positive head and neck cancer is associated with poorly differentiated, nonkeratinizing, basaloid morphology, wild-type p53, overexpression of p16, and inactivation of cyclin D1 and Rb [11, 24]. Table 3.1 summarizes the different characteristics of HPV-negative and HPV-positive OSCC [25].
Table 3.1
Differential features of HPV-positive and HPV-negative OSCC
|
Characteristics
|
HPV-positive OSCC
|
HPV-negative OSCC
|
|---|---|---|
|
Risk factor
|
Oral sex
|
Smoking, alcohol abuse
|
|
Age
|
Under 60 years
|
Above 60 years
|
|
Field carcinogenesis
|
Yes or no
|
Yes
|
|
Mutation of p53
|
Infrequently
|
Frequently
|
|
Susceptible site
|
Oropharynx
|
Various sites
|
|
Outcome
|
Good
|
Bad
|
3.4 Hallmarks of Cancer
Hanahan and Weinberg proposed six hallmarks of the cancer cell model : sustained proliferative signals, evasion of growth suppressors, resistance to cell death, replicative immortality, induction of angiogenesis , and activation of invasion and metastasis [26]. Recently, they added two emerging hallmarks, avoidance of immune destruction and deregulation of cellular energetics, and two enabling characteristics, genome instability and mutation and tumor-promoting inflammation (Fig. 3.2) [27]. Below, we describe OSCC-associated signals according to their recent model.
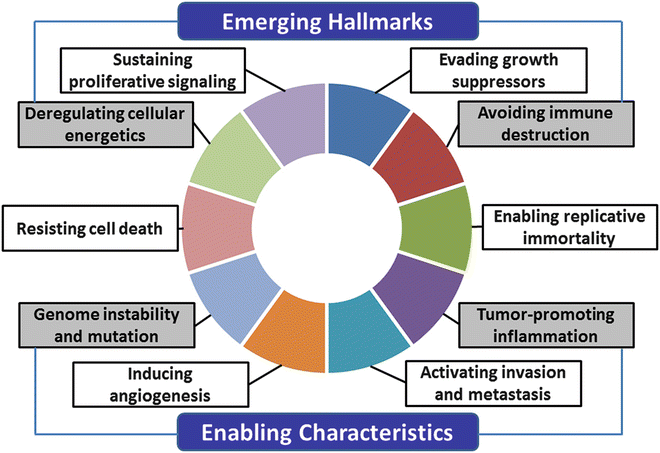
Fig. 3.2
The hallmarks of cancer. The new hallmarks of cancer were proposed by Hanahan et al. in 2011 [27]. This schema shows the ten hallmark capabilities including sustaining proliferative signals, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis and two emerging hallmarks, avoiding immune destruction, and deregulating cellular energetics and two enabling characteristics, genome instability and mutation and tumor-promoting inflammation
3.4.1 Sustained Proliferative Signals
Normal cells regulate growth signals through soluble and membrane-bound growth factors, but cancer cells are characterized by autonomous, chaotic growth because of the deregulation of growth signals.
3.4.1.1 EGFR
EGFR belongs to the membrane-bound receptor tyrosine kinase family comprising ErbB1 or human epidermal growth factor receptor 1 (HER 1), ErbB2 or HER2, ErbB3 or HER3, and ErbB4 or HER4 [28]. The binding of EGFR and EGF or transforming growth factor-α (TGFα) enhances Ras/Raf/mitogen-activated kinase protein (MAPK ), phosphatidylinositol-3-kinase (PI3K )/protein kinase B (AKT ), mammalian target of rapamycin (mTOR), Janus tyrosine kinase (JAK), signal transducers and activators of transcriptio n (STAT ), and protein kinase C (PKC) signaling pathways [12]. Overexpression of EGFR is observed in almost all patients with head and neck cancer and is correlated with advanced clinical stage and poor survival [29]. Additionally, overexpression of HER2 in head and neck squamous cell carcinoma (SCC) is correlated with nodal metastasis and poor outcome [30]. Cetuximab is an EGFR monoclonal antibody, and gefitinib and erlotinib are small-molecule EGFR tyrosine kinase inhibitors [12]. Molecular therapy targeting EGFR may be useful for OSCC.
3.4.1.2 Ras
The Ras family comprises H-Ras , K-Ras , and N-Ras in humans [31]. Approximately 30 % of all malignancies show mutations of Ras [32]. In advanced OSCC, a high frequency of H-Ras mutation is detected in Asian populations associated with betel nut chewing [33, 34]. On the other hand, mutations of H-Ras are not frequent (less than 6 %) in Western countries [35].
3.4.1.3 HGF /c-Met
Hepatocyte growth factor (HGF ) regulates cell growth, cell motility, and morphogenesis by activating tyrosine kinase signaling after binding to its receptor, c-Met [36]. In OSCC, HGF/c-Met promotes invasion and metastasis through the destruction of E-cadherin [37]. The expression rates of HGF in oral dysplasia and OSCC are higher than in non-tumor oral mucosa [38]. Through effects on matrix metalloproteinase (MMP )-1, MMP-2, and MMP-9 expression, overexpression of c-Met is correlated with nodal metastasis and poor outcome caused by enhanced migration and invasion [39]. Furthermore, c-Met induces lymphangiogenesis via potentiation of vascular endothelial growth factor (VEGF )-C [38].
3.4.1.4 NFκB
Nuclear factor-kappa B (NFκB ) is a ubiquitous nuclear transcription factor implicated in inflammation and the immune response [12, 32]. In its inactive state, NFκB is located in the cytoplasm bound to members of the NFκB inhibitor (IκB) family. After the phosphorylation, ubiquitination, and degradation of IκB, activated NFκB translocates to the nucleus and regulates the expression of target genes [40]. In OSCC, NFκB promotes lymphatic metastasis, invasion, and angiogenesis [41–43]. Moreover, the expression of NFκB has been shown to be related to resistance to chemoradiation therapy in OSCC [44].
3.4.1.5 PI3K -AKT
PI3K /AKT is a downstream target of EGFR that promotes the cell growth and proliferation of cancer cells [45]. PI3K is a heterodimeric kinase consisting of p85 and p110α (PI3KCA). Activation of PI3KCA and inactivation of phosphatase and tensin homolog (PTEN ) have been found in approximately 10 % of head and neck SCC [25, 46]. In OSCC, EGFR-independent activation of PI3K/AKT is frequently observed and strongly related to radiation resistance [45]. Furthermore, in their review article, Leemans et al. reported that the PI3K-PTEN-AKT pathway is one of the important signaling events in head and neck cancers, including OSCC [25].
3.4.1.6 Cyclin D1
Cyclin D1 is a cell cycle regulator from G1 to S phase. It interacts with calcium-dependent kinase 4 (CDK4) [17]. Overexpression of cyclin D1, found in 25–70 % of OSCC, is a nodal metastatic and poor prognostic factor [47, 48]. In signaling analysis using OSCC cells, the enhancement of extracellular signal-regulated kinase (ERK1/2) induces cyclin D1 expression [48]. Moreover, cyclin D1 is involved in cisplatin resistance through the repression of apoptosis and the activation of NFκB [49]. In HPV -related oral cancer, cyclin D1 activity is inhibited after the downregulation of p53 and pRb by E6 and E7, respectively [32].
3.4.1.7 STAT
Members of the STAT family are cytoplasmic transcription factors [12, 32]. STAT3 is activated by JAK or interleukin-6 (IL-6), EGF, platelet-derived growth factor (PDGF), VEGF , and others in several malignancies [50]. A recent study indicates that inactivation of STAT3 decreases hypoxia-inducible factor -1α (HIF-1 α) expression, inhibits cell growth, and augments the radiosensitivity of OSCC cells [51]. In immunohistochemical analysis using human OSCC specimens, pSTAT3 expression is correlated with poor prognosis and is observed in premalignant lesions [52]. Co-expression of STAT3 and c-Met is involved in OSCC progression [53].
3.4.2 Evasion of Growth Suppressors
Many tumor suppressor genes related to anti-growth signals are downregulated by mutation, deletion, and methylation in cancer cells. In the next sections, we review the functions of p53 , p16, p21, and PTEN in OSCC.
3.4.2.1 p53
The p53 gene is located on chromosome 17p13.1. p53 plays a pivotal role in the regulation of cell cycle, cellular differentiation, DNA repair, and apoptosis [12]. It is generally accepted that p53 is a gatekeeper of the genome [12]. Hypoxia, oncogenic stimulation by p14ARF activation, and DNA double-strand breaks increase p53 expression [54, 55]. Somatic mutations in p53 are detected in 60–80 % of head and neck cancers, including OSCC [25], and in 13 % of oral dysplasia [56]. Aside from mutations, the inactivation of p14ARF and the overexpression of mouse double minute 2 (MDM2) can also inhibit the tumor-suppressive function of p53 [12, 28]. Another mechanism for the deactivation of wild-type p53 is through high-risk HPV infection, such as HPV-16 and HPV-18. In HPV-positive OSCC, the inactivation of p53 occurs through the interaction with E6, which increases the ubiquitination and proteolysis of p53 [12, 57].
3.4.2.2 p16INK4A
The p16 tumor suppressor gene is located on chromosome 9p21-22. p16 disrupts the phosphorylation of Rb and induces the separation of E2F via binding with CDK4 and CDK6 [58]. Thus, p16 regulates the cell cycle from G1 to S phase [59]. Loss of p16 activation by methylation is frequently observed in OSCC, and p16 alteration is one of the initial events in oral carcinogenesis [60–62]. In contrast, Mendenhall et al. reported that high-risk HPV -related OSCC is associated with the overexpression of p16 [63].
3.4.2.3 p21WAF1
The cyclin-dependent kinase inhibitor p21 plays an essential role in regulating the G1-S transition of the cell cycle, cellular growth suppressors, and apoptosis [12, 32]. p21 is located downstream of p53 , and wild-type p53 induces p21 expression in response to DNA damage [64, 65]. The expression of p21 is observed not only in OSCC but also in the dysplastic lesions of the oral cavity; thus, activation of p21 is thought to be an early step in the malignant transformation of oral cancer [66]. Reduction of p21 levels is associated with poor outcome in OSCC [67]. However, other studies have shown that overexpression of p21 is correlated with poor prognosis [68]. In HPV -positive OSCC cases, p21 expression is a significant predictor of a favorable outcome [69]. Thus, the function of p21 in OSCC is controversial.
3.4.2.4 PTEN
The PTEN gene is located on chromosome 10q23.3 [70]. PTEN acts as a tumor suppressor through negative feedback of the PI3K /AKT pathway [71]. Deletion or somatic mutation of the PTEN gene has been detected in various carcinomas [70–74]. More recently, it was reported that reduction of PTEN is associated with poor prognosis. Therefore, the expression of PTEN may be a useful marker for radiotherapy in OSCC [75]. Kurasawa et al. reported that PTEN expression is downregulated in OSCC compared to the expression in normal oral mucosa [76]. They also observed restoration of PTEN mRNA in human OSCC-derived HSC3, HSC4, Ho-1-N-1, and SCC4 cells after treatment with 5-aza-2′-deoxycytidine (5-Aza-dc), a demethylation reagent. These results indicate that PTEN inactivation in OSCC may be due to gene methylation.
3.4.3 Resistance to Cell Death
Apoptosis, a programmed form of cell death, plays an important role in cellular homeostasis [12]. Apoptosis can occur through both extrinsic (receptor-initiated death) and intrinsic (mitochondrial) pathways. The extrinsic pathway is initiated by the binding of tumor necrosis factor-α (TNFα ) or Fas to TNF-related apoptosis-inducing ligand (TRAIL ) or Fas ligand (FasL ), leading to the activation of caspases 8 and 3. The intrinsic pathway is induced by the activation of BH3-containing proteins through various stimuli, prompting the release of cytochrome C and enhancement of caspases 9 and 3. Some important factors in apoptosis are the antiapoptotic proteins B-cell CLL/lymphoma 2 (Bcl-2 ) and Bcl-XL and the proapoptotic proteins Bax and Bak [12, 27, 32]. Figure 3.3 shows the process of apoptosis.
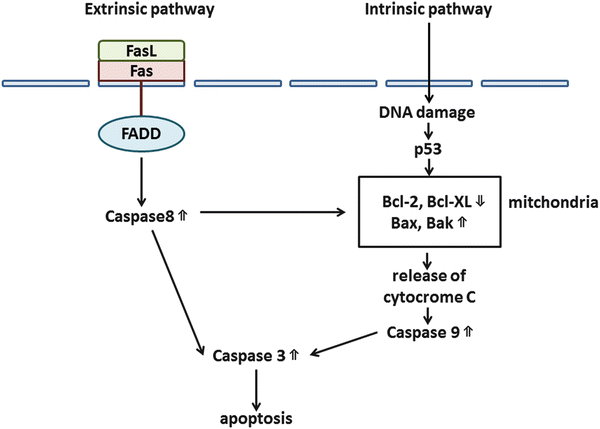
Fig. 3.3
Apoptotic pathways. There are two major programs of apoptosis: extrinsic and intrinsic pathways
3.4.3.1 Bcl-2 , Bcl-XL, Bax , and Bak
Previous reports suggested that the overexpression of Bcl-2 is a useful predictive marker of poor prognosis in OSCC [77]. Other reports linked the upregulation of Bcl-XL and wild-type p53 with cisplatin resistance in head and neck SCC cells [78]. Although Bax and Bak are proapoptotic proteins, previous studies demonstrated that their high expression is associated with poor prognosis in OSCC [79, 80]. However, another report showed that low expression of Bax is associated with poor prognosis [81]. Furthermore, decreased levels of Bax and Bak are found in hypoxia-induced apoptosis in OSCC [82].
3.4.4 Replicative Immortality
Telomeres are tandem repeats of the DNA sequence TTAGGG present at the linear ends of chromosomes. They become shortened in somatic cells [83]. The enzyme telomerase maintains telomere length. Its activity is regulated by the expression of human telomerase reverse transcriptase (hTERT ), the catalytic subunit of telomerase [12]. Activation of hTERT enables a cancer cell to maintain its telomeric length [32]. Recently, it was reported that hTERT stimulates epithelial-mesenchymal transition (EMT ) via the Wnt/β-catenin pathway and induces the stemness of cancer cells [84]. Telomerase or hTERT activity is found in 80 % of head and neck cancer, including OSCC [9]. Chen et al. reported that the activation of hTERT may be an early event of oral carcinogenesis, with high expression levels of hTERT related with poor outcome [85]. Furthermore, a recent report indicated that telomere dysfunction is a useful predictor of radioresistance in OSCC [86].
3.4.5 Induction of Angiogenesis
Angiogenesis and lymphangiogenesis are pivotal events in tumor progression and metastasis. The major angiogenic and lymphangiogenic factors are VEGF -A-VEGF receptor -1/2 (VEGFR -1/2) and the VEGF-C/D-VEGFR-3 system, respectively [87]. We recently showed that high microvessel density (MVD) and lymphovessel density (LVD) are strongly associated with T grade, clinical stage, nodal metastasis, local recurrence, and poor outcome in OSCC [88]. Other important factors for angiogenesis and lymphangiogenesis include basic fibroblast growth factor (bFGF) [89], interleukin (IL)-8 [90], platelet-derived growth factor beta polypeptide (PDGFB) [89], angiopoietin2 [91], FoxC2 [92], Prox1 [93], and neuropilin2 [94].
3.4.5.1 VEGF Family
The VEGF family includes VEGF-A, VEGF-B, placenta growth factor (PlGF), VEGF-C, VEGF-D, and VEGF-E and plays a key role in developmental vasculogenesis and inflammatory angiogenesis [87]. We performed immunohistochemistry of VEGF family using OSCC samples and found that VEGF-A expression was related to tumor progression, lymph node metastasis, disease recurrence, and poor prognosis; the expression of VEGF-C and VEGF-D was only linked to nodal metastasis. Furthermore, VEGF-D was induced only in lymphangiogenesis , whereas VEGF-A and VEGF were induced in both angiogenesis and lymphangiogenesis in OSCC [88]. The recently developed drug bevacizumab is an anti-VEGF-A agent, and it may be useful for the treatment of OSCC [95].
3.4.6 Activation of Invasion and Metastasis
The following steps are necessary for cancer cell invasion and metastasis: destruction of the basal membrane, decreased adhesion and isolation of cancer cells, acquisition of cancer cell migration and stromal infiltration, vascular invasion, intravascular migration, and cancer cell growth in the metastatic organ. Here, we review E-cadherin , MMP s, and integrins.
3.4.6.1 E-Cadherin
E-cadherin , a calcium-dependent intracellular adhesion molecule, plays an important role in maintaining cell-to-cell connections in normal epithelial cells [96]. In OSCC, reduction of E-cadherin is associated with invasion [97], nodal metastasis [96], and poor prognosis [98]. Inactivation of E-cadherin can be induced by methylation [99]. Its transcriptional inhibition results from the activation of Snail, Slug, zinc finger E-box binding homeobox 2 (ZEB2), and Twist [100]. Thus, E-cadherin plays a pivotal role in EMT .
3.4.6.2 MMP s
MMP s are extracellular proteases involved in extracellular matrix modulation [12, 32]. Many studies have described the relationship between MMPs and OSCC. MMP-2 is a good predictive marker of invasion and metastasis in OSCC [101]. The high expression of MMP-7 has been linked to invasion and poor prognosis [102]. MMP-3, MMP-9, MMP-10, and MMP-11 may also take part in the progression of OSCC [103–106]. Tissue inhibitors of metalloproteinase (TIMP), antagonists of MMPs, are associated with the progression of OSCC [105, 107]. However, the relationship between TIMP/MMPs and clinical features is controversial.
3.4.6.3 Integrin s
Integrin s are heterodimeric cellular transmembrane proteins that mediate cell-to-cell and cell-to-extracellular matrix interactions [32]. In OSCC, integrins α3β1 and α6Aβ1 are related to invasion, metastasis, and poor prognosis [108]. The interaction of integrin α5 and hypoxia-inducible factor-1 (HIF-1 α) promotes the invasion of OSCC cells under hypoxic conditions [109]. Recently, Li et al. revealed that the overexpression of integrin αvβ6 is related to unfavorable clinical prognostic factors and decreased survival [110]. The upregulation of integrin αvβ6 is also found in oral dysplasia [111]. However, the role of integrins in cancer is complicated, and further studies are needed.
3.4.7 Avoidance of Immune Destruction
The function of immune cell cancer is an emerging hallmark of cancer [27]. The marked infiltration of CD8+ cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells is related to a favorable outcome in colon and ovarian cancers [27, 112, 113]. A previous report suggested that TGFα secreted by cancer cells confuses infiltrating CTLs and NK cells [114], whose activation is also attenuated by tumor-infiltrating myeloid cells [115]. Toll-like receptors (TLRs ) are single transmembrane cell-surface receptors with tumor immune escape and resistance to apoptosis properties [116]. In OSCC, TLR3, 4, 7, and 9 are correlated with invasion, apoptosis, necrosis, and cell proliferation [117–120].
3.4.8 Tumor-Promoting Inflammation
Inflammation can change the tumor microenvironment by inducing growth factors and modifying the extracellular matrix, thus facilitating angiogenesis , invasion, and metastasis [27]. Furthermore, chronic inflammation increases cancer risk because activated inflammatory cells release reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI) and induce DNA damage and genomic instability [121]. Therefore, it is now believed that chronic inflammation promotes tumor progression.
3.4.8.1 COX-2
COX-2 is a proinflammatory enzyme that converts arachidonic acid to prostaglandins. Despite the lack of COX-2 expression in normal epithelium, increased expression is found in OSCC and is related to apoptosis inhibition [28, 32]. Overexpression of COX-2 is found in higher grades of oral epithelial dysplasia and in invasion and nodal metastasis of OSCC [122–124]. Additionally, COX-2 inhibition suppresses proliferation, migration, and invasion in OSCC cells [125].
3.4.9 Genome Instability and Mutation
Epigenetic mechanisms, such as DNA methylation and histone deacetylation , play pivotal roles in silencing tumor suppressor genes. One of the famous methylated genes in OSCC is CDKN2A, which controls the cell cycle [126, 127]. Loss of E-cadherin (CDH1 gene) is observed in 64 % of primary OSCC, 67 % of nodal metastases, and 71 % of recurrent tumors [128]. Furthermore, the DNA repair genes O6-methylguanine-DNA-methyltransferase (MGMT) and death-associated protein kinase (DAPK) were methylated in 52 % and 68 % of OSCCs, respectively [129]. Retinoic acid receptor-β2 (RAR-β2) is a member of the thyroid-steroid hormone receptor superfamily of nuclear transcriptional regulators. Youssef and colleagues reported that RAR-β2 methylation is found in 66 % of head and neck SCC, including OSCC, and 53 % of oral leukoplakia [130].
3.4.10 Deregulation of Cellular Energetics
Under aerobic conditions, normal cells produce adenosine triphosphate (ATP) from glucose through the tricarboxylic acid (TCA) cycle in the mitochondria and oxidative phosphorylation in the electron transfer system. In addition, ATP is also produced through the anaerobic glycolytic pathway, which breaks down glucose in the cytoplasm and generates lactic acid under anaerobic conditions. However, cancer cells can produce ATP through aerobic glycolysis even in the presence of oxygen. This phenomenon is called the Warburg effect [27]. In clinical settings, glucose metabolism in tumors is assessed by positron emission tomography (PET) using 18F-fluorodeoxyglucose (FDG).
3.4.10.1 GLUT-1
3.4.10.2 HIF-1
HIF-1 is composed of HIF-1α, which modulates the transactivation of target genes under hypoxic conditions by binding to hypoxia-responsive elements, and HIF-1β, a key regulator of oxygen homeostasis [82, 134]. HIF-1α plays an important role in angiogenesis by activating VEGF -A and inhibiting apoptosis; it is associated with poor prognosis in OSCC [82, 135–137]. Moreover, HIF-1α promotes nodal metastasis by the induction of lymphangiogenesis via VEGF-C [138].
3.5 EMT
EMT , characterized by the loss of epithelial differentiation and the acquisition of a mesenchymal phenotype, plays a key role in cancer metastasis [139]. The important factors for EMT are TGFβ, Wnt, Notch, interleukin-like EMT inducer (ILEI), HGF , EGF, and PDGF. TGFβ acts as tumor suppressor by inhibiting cell growth and facilitating apoptosis, but it has tumor-promoting effects by inducing EMT in advanced cancers [140]. Cancer cells with induced EMT display a loss of epithelial cell-to-cell contacts, with suppression of E-cadherin , ZO-1, and claudin-1/-7, among others, and expression of mesenchymal markers such as α-smooth muscle actin (SMA), vimentin, N-cadherin, and desmin [140, 141]. In cancer cells, the upregulation of transcription factors such as Snail, Slug, Twist, ZEB1/2, and/or E47 is necessary for the maintenance of EMT [141]. The induction of EMT is also critical for the progression of OSCC [139, 142, 143].
3.6 MicroRNA
MicroRNAs (miRNAs) are noncoding small RNAs of approximately 18–25 nucleotides that regulate gene expression by binding to the 3′-untranslated region (UTR) of target mRNAs [144]. Mature miRNAs derive from primary miRNAs (pri-miRNAs), which are processed in the nucleus into precursor miRNAs (pre-miRNAs) by the RNase Drosha and its cofactor DiGeorge syndrome critical region gene 8 (DCRG8). The pre-miRNAs are exported to the cytoplasm by exportin-5 and processed into mature miRNAs by the RNase Dicer. Once integrated into the RNA-induced silencing complex (RISC), the mature miRNAs mediate target gene mRNA expression (Fig. 3.4) [145–148]. Here, we summarize the role of several miRNAs in OSCC [146, 147].
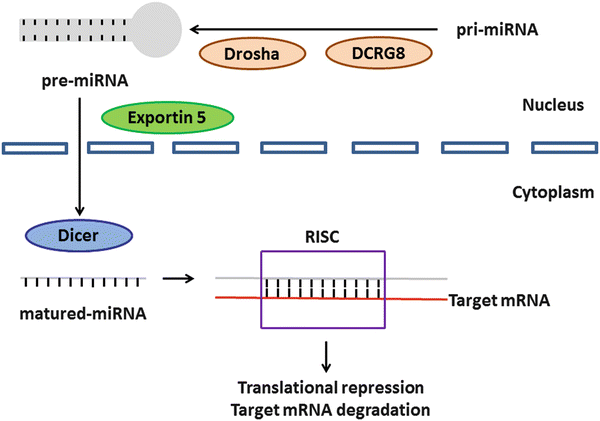
Fig. 3.4
The cascade of miRNA in humans. This schema was modified from previous findings [147, 148]. The pri-miRNAs are processed in the nucleus into pre-miRNAs with the help of Drosha and its cofactor DCRG8. The pre-miRNAs are exported to the cytoplasm by exportin-5 and processed into mature miRNAs by another Dicer. The mature miRNAs are integrated into the RISC and regulate target gene mRNA expression
3.6.1 Upregulation of miRNAs in OSCC
Overexpression of miR-21, which targets tropomyosin 1 (TPM1) and PTEN , is related to the reduction of apoptosis [146, 147, 149]. Similarly, upregulation of miR-24 reduces apoptosis by inhibiting RNA-binding protein dead end 1 (DND1) and promotes proliferation through the downregulation of cyclin-dependent kinase inhibitor 1B (CDKN1B), a downstream target of DND1 [146, 147, 150]. Elevated expression of miR-222 inhibits cell invasion by directly regulating MMP -1 and indirectly reducing manganese superoxide dismutase 2 (SOD2) expression [146, 147, 151]. Another report showed that high expression of miR-211 is correlated with nodal metastasis, vascular invasion, and poor prognosis and that the increment of miR-211 expression results in the attenuation of α-galactosidase and the LacZ operon [146, 147, 152]. The upregulation of miR-31 directly represses factor-inhibiting hypoxia-inducible factor (FIH) expression. The plasma level of miR-31 in patients with OSCC is higher than that in controls [146, 147, 153, 154]. Moreover, overexpression of miR-23a and miR-98 suppresses the expression of topoisomerase IIβ (TOP2B) that is an inhibitor of TOP2 and high-mobility group box A2 (HMGA2) that is an enhancer of cisplatin and doxorubicin resistance, respectively [146, 147, 155, 156].
3.6.2 Downregulation of miRNAs in OSCC
Underexpression of miR-133a elevates the expression of pyruvate kinase type M2 (PKM2) and glutathione-S-transferase P1 (GSTP1) and is associated with promotion of cell proliferation and inhibition of apoptosis [146, 147, 157, 158]. Liu et al. revealed that low miR-138 expression affects metastasis by directly targeting Rho-related GTP-binding protein C (RhoC) and Rho-associated, coiled-coil-containing protein kinase 2 (ROCK2) [146, 147]. miR-15a is also downregulated in OSCC, resulting in regulation of PKCα and cyclin E expression, which in turn affects DNA synthesis [146, 147, 158]. Furthermore, diminished expression of miR-7 and miR-124, which target insulin-like growth factor 1 (IGF1) and integrin beta-1 (ITGB1), respectively, is observed in OSCC [147, 159, 160].
3.7 The Novel OSCC-Related Molecular Markers
3.7.1 RAGE -HMGB1 Signal
Receptor for advanced glycation end products (RAGE ) is a multi-ligand receptor that belongs to the immunoglobulin superfamily [161]. High-mobility group box 1 (HMGB1 ), AGE, S100 protein, and β-amyloid are major ligands of RAGE, whose associated intracellular signaling pathways involve GTPases, Rac1/Cdc42, MAPK , ERK1/2, c-Jun N-terminal kinase (JNK), and NFκB [162]. HMGB1 is a nuclear chromatin protein and cytokine that participates in inflammation and cancer progression [163]. Extracellular HMGB1 is associated with endotoxin-induced tissue injury, macrophage infiltration, and tumor advancement [163, 164]. We previously reported that the RAGE-HMGB1 system is deeply correlated with tumor progression, metastasis, prognosis of gastrointestinal cancer [161–163], prostate cancer [165], and malignant transformation of colorectal adenoma [166].
The RAGE -HMGB1 system also plays an important role in OSCC. Increased expression of RAGE was linked to invasion depth and poor prognosis, and multivariate analysis demonstrated a significant relationship between RAGE expression levels and disease-free survival [167]. Furthermore, RAGE-HMGB1 signaling induces angiogenesis by activation of VEGF -A [168], and it is responsible for migration, invasion, and MMP -9 production in OSCC cells [169]. Moreover, RAGE is involved in rat tongue carcinogenesis induced by 4-nitroquinoline 1-oxide (4-NQO) [170]. Interestingly, a selective COX-2 inhibitor, etodolac, significantly reduced the expression of RAGE in dysplasia and OSCC. Our findings suggest that the RAGE-HMGB1 system might be a good marker for malignancy potential in OSCC and a useful target for the diagnosis and treatment of OSCC.
3.7.2 MIA Gene Family
The melanoma inhibitory activity (MIA ) gene family contains MIA, MIA2, MIA3, and otoraplin (OTOR), which share 47–59 % cDNA sequence and 34–45 % amino acid homology [171]. Previous reports indicated that MIA promotes the progression and metastasis of malignant melanoma and other malignancies [172, 173]. MIA accelerates the cell detachment, migration, invasion, apoptosis resistance, and infiltration of lymphokine-activated killer cells [174]. MIA inhibits cell-to-stromal interaction by binding to fibronectin via SH3 domain-like structures [174, 175]. Bauer et al. also revealed that MIA is a ligand for selected integrins by demonstrating its ability to bind integrins α4β1 and α5β1 [176]. Recently, we elucidated the functional role of MIA in OSCC [177, 88]. The interplay between intracellular HMGB1 and NFκB p65 facilitated MIA expression. MIA also regulated the phosphorylation of ERK1/2 and MAPK p38, the induction of angiogenesis and lymphangiogenesis , and the activation of VEGF -A/-C/-D in OSCC cells. Analysis of OSCC specimens revealed that MIA expression was strongly related with nodal metastasis, poor prognosis, MVD, LVD, and high expression levels of HMGB1 and VEGF family members. Figure 3.5 summarizes the roles of MIA in OSCC.
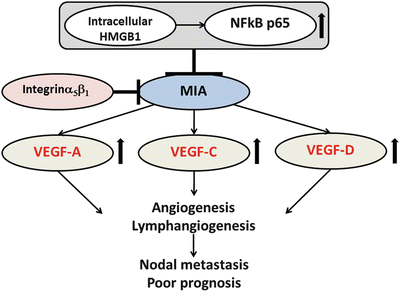
Fig. 3.5
The schema of the roles of MIA in OSCC. MIA acts as oncogene by induction of angiogenesis and lymphangiogenesis via activation of VEGF family and is associated with nodal metastasis and poor prognosis in OSCC
The activation of MIA 2 is transcriptionally regulated by the hepatocyte nuclear factor (HNF)-1 binding site [178]. Although MIA2 expression is increased chronic hepatitis and liver cirrhosis [179], MIA2 inhibits the growth and invasion of hepatocellular carcinoma (HCC) [180]. In OSCC, MIA2 expression was strongly correlated with tumor progression and nodal metastasis and was negatively associated with cytotoxic T lymphocyte infiltration [181]. In in vitro examinations using OSCC cells, modulation of cancer cell invasion, antiapoptotic effect, VEGF family expression, and host anticancer immunity were found to be related to MIA2. Moreover, MIA2 regulated the phosphorylation of MAPK p38 and JNK by binding to integrins α4 and α5.
MIA 3 acts as tumor suppressor in melanoma, colorectal cancer, and HCC [182, 183]. We also showed that it induces angiogenesis and lymphangiogenesis in OSCC (unpublished data). OTOR expression is highly restricted in healthy eyes, cochlea, and cartilage [184], and its involvement in cancer has not been reported yet. Our results suggest that the MIA gene family might be important for tumor progression in OSCC.
3.7.3 Trk
Tropomyosin receptor kinases (Trk) have a high affinity for neurotrophins. Nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) or neurotrophin 4/5 (NT4/5), and NT3 are the ligands for TrkA , TrkB , and TrkC , respectively [185]. Trks regulate neuronal survival and differentiation [186]. Trks also act as oncogenes in many tumors, and the overexpression of TrkA is found in thyroid and ovarian cancer [187, 188]. Overexpression of TrkB has been found in hepatocellular carcinoma [189], pancreatic cancer [190], and neuroblastoma [191]; it is correlated with more aggressive tumor behavior. TrkB and TrkC also inhibit apoptosis in ovarian cancer and neuroblastoma cells, respectively, by activating PI3K signaling [192, 193]. Furthermore, we showed that TrkB and TrkC have a tumor progression function in colorectal cancer [194]. However, it has been reported that TrkB is downregulated in prostate cancer [195]. In neuroblastoma, patients with high expression of TrkA or TrkC have a better prognosis [196, 197], and TrkC plays a favorable role in medulloblastoma, leading to a good clinical outcome [198]. Therefore, the detailed functional roles of Trks in malignancies are not clearly understood.
We recently reported the functional roles of Trks in OSCC [199]. Although all Trks inhibited apoptosis by activating caspase 3, regulation of cell growth by TrkB – and MMP -2/-9-mediated enhancement of invasive ability by TrkC was observed. The gene expression of Trks was modulated by methylation of runt-related transcription factor 3 (RUNX3 ). VEGF family-dependent and VEGF family-independent angiogenesis and lymphangiogenesis were induced by TrkB and TrkC, respectively. In immunohistochemical analysis using OSCC specimens, the expression of TrkB was significantly associated with MVD, LVD, and poor outcome, whereas the immunoreactivity of TrkC was strongly correlated with clinical stage, nodal metastasis, MVD, LVD, and poor prognosis. These results indicate that Trks might be a useful diagnostic and therapeutic tool for OSCC. The functional roles of Trks in OSCC are summarized in Fig. 3.6.
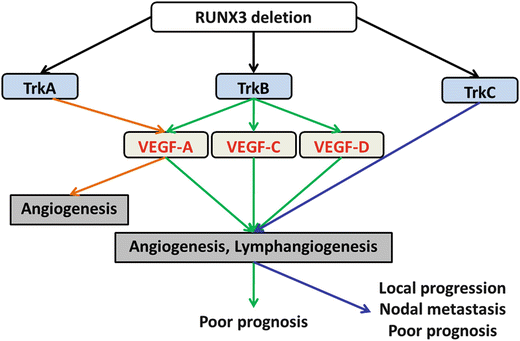
Fig. 3.6
The schema of the function of Trk in OSCC. Trks are regulator of angiogenesis and lymphangiogenesis and promote tumor progression, nodal metastasis, and poor outcome of OSCC
3.7.4 miR-126
miR-126 is an endothelial-specific miRNA that is located within intron 7 of epidermal growth factor-like domain 7 (EGFL7 ). Its overexpression in endothelial cells enhances VEGF -A activity and promotes vessel formation by repressing the expression of sprouty-related protein-1 (Spred-1) in developmental angiogenesis and vasculogenesis in wound healing [200, 201]. Low expression of miR-126 is found in pancreatic cancer, where it is associated with the activation of K-Ras [202], and it is significantly associated with short survival in non-small cell lung carcinoma (NSCLC) and renal cell carcinoma [203, 204]. Downregulation of miR-126 and EGFL7 by DNA hypermethylation has been reported in urologic cancer and NSCLC [205, 206]. However, miR-126 promotes gastric carcinogenesis and metastasis in prostatic adenocarcinoma [207, 208]. Thus, the functional role of miR-126 in cancer is still unknown. We clarified that miR-126 negatively regulates VEGF-A activation and promotes cell proliferation. Demethylation upon 5-Aza-dc treatment increased the expression of miR-126 and EGFL7 in OSCC cells [209]. A significant relationship was also found between decreased expression of miR-126 and tumor progression, nodal metastasis, induction of angiogenesis/lymphangiogenesis , and poor prognosis in 118 OSCC cases. Moreover, low expression of miR-126 was a prognostic factor for disease-free survival according to multivariate analysis. Our results imply that the restoration of miR-126 expression could be a useful therapeutic target for human OSCC.
3.8 Salivary Gland Cancer
Salivary gland cancer is a rare tumor whose estimated annual incidence in Japan is approximately 400 cases [210]. The expression of HER 2 [211] and the downregulation of E-cadherin [212] and p27 [213] have been reported to promote tumor progression in ACC. Cyclic adenosine monophosphate response element binding protein (CREB)-regulated transcription coactivator 1-mastermind-like gene family (CRTC1-MAML2) fusion oncogene is a good marker for MEC [214]. However, the useful molecular markers associated with tumor progression of ACC and MEC remain controversial. Below, we describe our findings concerning ACC and MEC, the representative salivary gland cancers in Japan.
3.8.1 Reg IV
Regenerating islet-derived family member 4 (Reg IV ), a member of the Reg gene family, belongs to the calcium-dependent superfamily. Its expression is confined to the gastrointestinal tract and pancreas in normal tissue, and it was detected in Crohn’s disease, ulcerative colitis, and the intestinal metaplasia of gastric mucosa [215]. In malignancies, upregulation of Reg IV is found in gastric, colorectal, pancreatic, and prostate cancer [216–219]. We recently reported that Reg IV accelerates peritoneal metastasis and is detected in lavage fluids in patients with gastric adenocarcinoma [220]. We also revealed that 41 % of ACC expresses Reg IV and that overexpression of Reg IV is strongly associated with tumor recurrence and poor prognosis [221]. Furthermore, we reported that Reg IV induces EGFR phosphorylation and promotes cell growth in ACC. Reg IV has been observed in intestinal-type gastric cancer cells that express the intestinal mucin marker MUC2 [216]; however, no relationship was found between Reg IV and MUC2 expression in ACC. Thus, Reg IV might not be related to the specific differentiation of salivary gland epithelial cells. Our results suggest that Reg IV will be a good marker for poor prognosis in salivary gland ACC.
3.8.2 RUNX3
RUNX3 is a transcription factor that belongs to the TGFβ superfamily [222]. RUNX3 possesses tumor-suppressive functions, and its expression is decreased or abolished by CpG island hypermethylation in its promoter region in several tumors [223]. Additionally, cytoplasmic mislocalization of RUNX3 is associated with inactive tumor suppressor function [224]. We described the expression and methylation status of RUNX3 in salivary gland ACC and MEC [225]. Using immunohistochemical analysis, we showed that RUNX3 was mislocalized, decreased, or absent in all cases examined and that the decrease or deletion of RUNX3 was significantly correlated with tumor progression and short disease-free periods in ACC and MEC. Furthermore, by methylation-specific PCR and bisulfite genomic sequencing using microdissected cDNA, low mRNA expression and enhanced DNA hypermethylation of RUNX3 were found in ACC and MEC, when compared to benign salivary gland tumor and normal salivary gland. These results indicate that restoring the tumor-suppressive ability of RUNX3 may be a useful therapeutic target in salivary gland ACC and MEC.
3.9 Conclusions
The molecular mechanisms of carcinogenesis, tumor progression, and metastasis of head and neck cancer have been elucidated by recent advances in molecular biology. However, many unsolved questions remain. Current understanding of pathology is based on morphological findings. However, the molecular pathological approach is becoming mainstream in current pathological research because pathologists are increasingly required to master molecular biological knowledge. Our future efforts will focus on the elucidation of the morphology and molecular pathology of oral cancer.
References
1.
Argiris A, Karamouzis MV, Raben D, Ferris RL (2008) Head and neck cancer. Lancet 371(9625):1695–1709. doi:10.1016/s0140-6736(08)60728-x PubMed
2.
Paterson IC, Eveson JW, Prime SS (1996) Molecular changes in oral cancer may reflect aetiology and ethnic origin. Eur J Cancer Pt B Oral Oncol 32B(3):150–153
3.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90. doi:10.3322/caac.20107 PubMed
4.
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62(1):10–29. doi:10.3322/caac.20138 PubMed
5.
Tanaka S, Sobue T (2005) Comparison of oral and pharyngeal cancer mortality in five countries: France, Italy, Japan, UK and USA from the WHO Mortality Database (1960–2000). Jpn J Clin Oncol 35(8):488–491. doi:10.1093/jjco/hyi133 PubMed
6.
Alvi A, Johnson JT (1997) Development of distant metastasis after treatment of advanced-stage head and neck cancer. Head Neck 19(6):500–505PubMed
7.
Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper C, Jerjes W, Upile T, Kalavrezos N, Violette SM, Weinreb PH, Chester KA, Chana JS, Marshall JF, Hart IR, Hackshaw AK, Piper K, Thomas GJ (2011) Stromal features are predictive of disease mortality in oral cancer patients. J Pathol 223(4):470–481. doi:10.1002/path.2830 PubMed
8.
Izumo T, Kirita T, Ariji E, Ozeki S, Okada N, Okabe S, Okazaki Y, Omura K, Kusama M, Sato T, Shinohara M, Shimozato K, Shintani S, Tanaka Y, Nakayama E, Hayashi T, Miyazaki A, Yagishita H, Yamane M, Working Group 1 on the Guidelines for C, Pathological Studies of Oral Cancer SCJSfOT (2012) General rules for clinical and pathological studies on oral cancer: a synopsis. Jpn J Clin Oncol 42(11):1099–1109. doi:10.1093/jjco/hys141 PubMed
9.
Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D (1996) Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res 56(11):2488–2492PubMed
10.
Lippman SM, Sudbo J, Hong WK (2005) Oral cancer prevention and the evolution of molecular-targeted drug development. J Clin Oncol 23(2):346–356. doi:10.1200/jco.2005.09.128 PubMed
11.
Perez-Ordonez B, Beauchemin M, Jordan RC (2006) Molecular biology of squamous cell carcinoma of the head and neck. J Clin Pathol 59(5):445–453. doi:10.1136/jcp.2003.007641 PubMedCentralPubMed
12.
Choi S, Myers JN (2008) Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dental Res 87(1):14–32
13.
Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W, Ahrendt S, Eby Y, Sewell D, Nawroz H, Bartek J, Sidransky D (1996) High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 56(16):3630–3633PubMed
14.
Garnis C, Baldwin C, Zhang L, Rosin MP, Lam WL (2003) Use of complete coverage array comparative genomic hybridization to define copy number alterations on chromosome 3p in oral squamous cell carcinomas. Cancer Res 63(24):8582–8585PubMed
15.
Shahnavaz SA, Regezi JA, Bradley G, Dube ID, Jordan RC (2000) p53 gene mutations in sequential oral epithelial dysplasias and squamous cell carcinomas. J Pathol 190(4):417–422. doi:10.1002/(sici)1096-9896(200003)190:4<417::aid-path544>3.0.co;2-g PubMed
16.
Boyle JO, Hakim J, Koch W, van der Riet P, Hruban RH, Roa RA, Correo R, Eby YJ, Ruppert JM, Sidransky D (1993) The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res 53(19):4477–4480PubMed
17.
Yu Z, Weinberger PM, Haffty BG, Sasaki C, Zerillo C, Joe J, Kowalski D, Dziura J, Camp RL, Rimm DL, Psyrri A (2005) Cyclin d1 is a valuable prognostic marker in oropharyngeal squamous cell carcinoma. Clin Cancer Res 11(3):1160–1166PubMed
18.
Sudbo J, Kildal W, Risberg B, Koppang HS, Danielsen HE, Reith A (2001) DNA content as a prognostic marker in patients with oral leukoplakia. New Engl J Med 344(17):1270–1278. doi:10.1056/nejm200104263441702 PubMed
19.
Sudbo J, Lippman SM, Lee JJ, Mao L, Kildal W, Sudbo A, Sagen S, Bryne M, El-Naggar A, Risberg B, Evensen JF, Reith A (2004) The influence of resection and aneuploidy on mortality in oral leukoplakia. New Engl J Med 350(14):1405–1413. doi:10.1056/NEJMoa033374 PubMed
20.
Kreimer AR, Clifford GM, Boyle P, Franceschi S (2005) Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev 14(2):467–475. doi:10.1158/1055-9965.epi-04-0551 PubMed
21.
Adelstein DJ, Ridge JA, Gillison ML, Chaturvedi AK, D’Souza G, Gravitt PE, Westra W, Psyrri A, Kast WM, Koutsky LA, Giuliano A, Krosnick S, Trotti A, Schuller DE, Forastiere A, Ullmann CD (2009) Head and neck squamous cell cancer and the human papillomavirus: summary of a National Cancer Institute State of the science meeting, November 9–10, 2008, Washington, DC. Head Neck 31(11):1393–1422. doi:10.1002/hed.21269 PubMed
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


