Introduction
Mandibular growth is believed to be strongly related to mastication. Furthermore, mandibular condylar cartilage is known to be derived from neural crest cells. We examined whether the degree of chewing affects condylar cartilage growth of the mandible.
Methods
Mice were fed diets with varying hardness. Genes specific to neural crest–derived cells were measured by real-time polymerase chain reaction to compare the expression changes between the mandibular and tibia cartilages. The mandibular condylar cartilage was then evaluated histologically, and proliferation was evaluated using proliferating cell nuclear antigen. Immunostaining was conducted for osteopontin, type X collagen, and Musashi1, and real-time polymerase chain reaction was used to assess the expression levels of osteopontin and type X collagen.
Results
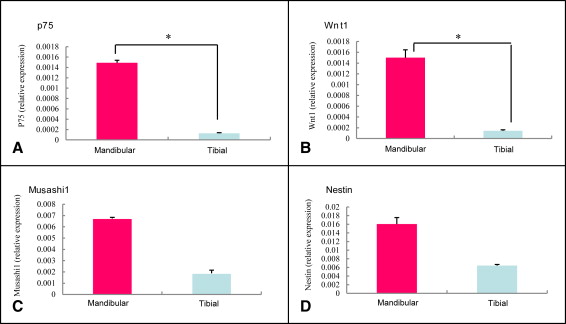
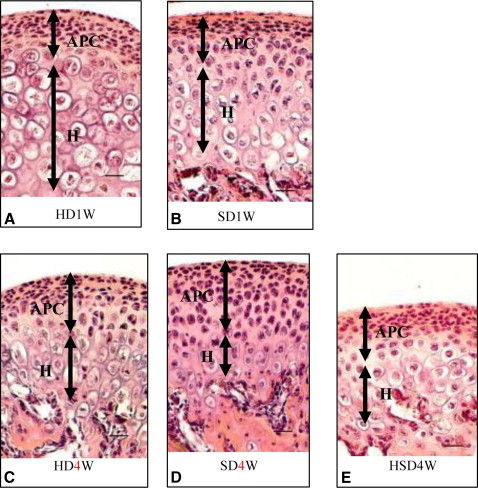
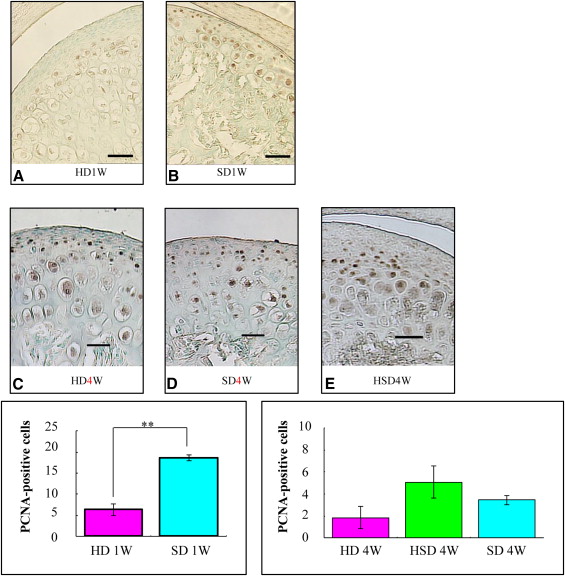
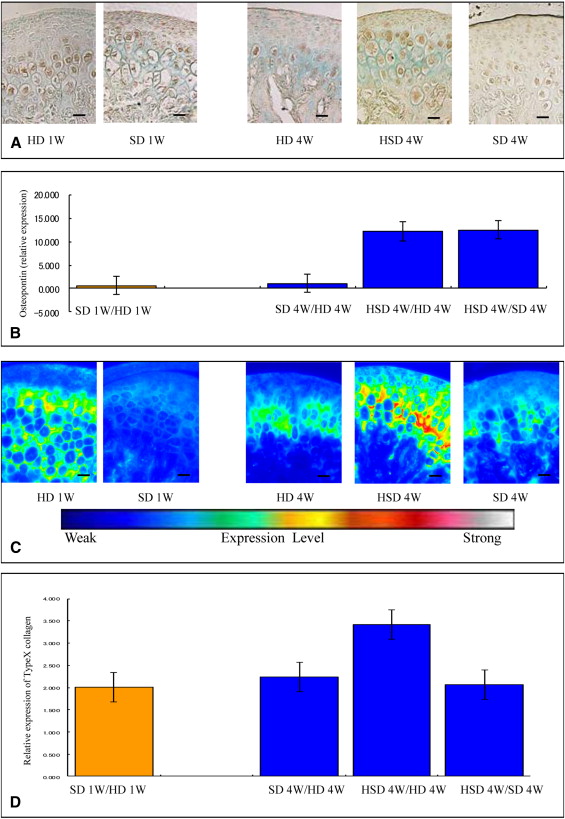
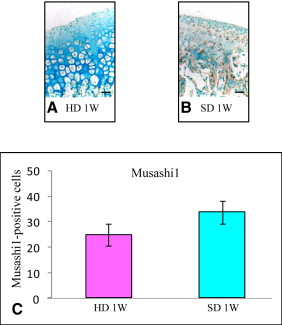
Markers including P75, Wnt-1, Musashi1, and Nestin were upregulated in the mandibular condylar cartilage as compared with the tibial cartilage. Histologic assessment of the mandibular cartilage showed that the hypertrophic chondrocyte zone was statistically significantly thicker in mice fed a hard diet. Chondrocyte proliferation and Musashi1 expression were lower in mice fed a hard diet. After 4 weeks, numerous osteopontin and type X collagen-positive cells were observed in mice fed a mixed diet.
Conclusions
Mastication affects the balance between differentiation and proliferation in the mandibular condylar cartilage. This phenomenon might be attributed to the presence of neural crest–derived cells.
Highlights
- •
Mastication markedly affects mandibular condylar cartilage growth in rodents.
- •
Food consistency influences morphologic and histologic characteristics of mandibular growth.
- •
Neural crest-derived cells might help mandibular condylar cartilage to adapt.
Endochondral ossification of the mandibular condylar cartilage is thought to occur at the growth center site of the mandible, and the mandibular condyle has a high ability to adapt. It is considered that mandibular condylar cartilage is more sensitive to changes in mechanical conditions than other types of cartilage, and that muscular function around the jaw has a marked influence on maxillofacial growth. The mandibular cartilage is thought to consist of cells originating from the neural crest that are multipotential with a high proliferative potency. However, it is unknown whether these characteristics are observed in vivo.
We previously reported that changes in mastication markedly influenced the expression of several genes. Using laser microdissection, along with cDNA array and real-time polymerase chain reaction (PCR) techniques, we showed that it was possible to accurately quantify gene expression levels in vivo by selectively obtaining cells from the mandibular condylar cartilage, and identified the expression levels of several genes that were significantly altered between the lactation and mastication periods. Therefore, we hypothesized that mastication would influence condylar cartilage responses and subsequent growth of the mandible. It is known that maxillofacial structures change with different diets, with the ramus height reported to be higher in rats fed a variable diet. The mandibular condylar cartilage also undergoes morphologic changes with different diets, since it was found to be thicker and highly proliferative in rats fed a soft diet. Altered functional loading caused a decrease in the expression of chondrocyte differentiation markers and a decrease in proteoglycan staining in the mandibular condylar cartilage. Total RNA expression analyses of condyles from mice fed soft food showed a significant decrease in the osteopontin levels. Furthermore, accelerated degeneration of the condylar cartilage led to its enhanced replacement with bone, resulting in thinner cartilage. Compressive forces have also been reported to affect the mitotic activity of chondrocytes, and thereby further affect the cell layer thickness.
Although a number of studies have investigated the effects of mastication on the growth of the mandible, there have been no cohesive reports investigating the adaptive ability of the mandibular condylar cartilage and the expression levels of genes related to mandibular condylar cartilage growth, and the 3-dimensional shape of the mandible in relation to mastication. In our previous study, we investigated the effects of mastication on mandibular morphology as assessed by microcomputed tomography, and found significantly thicker condylar widths in mice fed a hard-pellet diet (HD) for 1 week than in mice fed a soft diet (SD). Interestingly, mandibular length was greatest in mice fed a combined hard and soft diet (HSD; 2 weeks of each) and smallest in the HD group. Ramus height was greatest in the HSD group and lowest in the SD group, and bone volume was also lower in the SD group than in the HSD and HD groups. However, the adaptive mechanism of the mandibular condylar cartilage has remained unclear.
In this study, we first used real-time PCR to examine the mandibular cartilage gene expression differences from that of other regions, using tibial cartilage as a comparison. Next, we investigated the causes of regeneration in the composition of the mandibular condylar cartilage and assessed whether changes in the level of mastication influenced mandibular growth using histologic and molecular analyses. Furthermore, we examined the protein and mRNA expression levels of osteopontin and type X collagen as well as the protein expression of Musashi1 in the mandibular condylar cartilage by immunostaining.
Material and methods
Forty-eight 21-day-old male CD-1 mice (Saitama Experimental Animals Supply, Saitama, Japan) were used. The mice were randomly divided into 6 groups to receive (ad libitum) diets of varying hardness and durations: control (3 animals), HD/1 week (9 animals), HD/4 weeks (9 animals), SD/1 week (9 animals), SD/4 weeks (9 animals), and HSD/4 weeks (9 animals). The 3 control mice were used to generate Figure 1 , whereas 3 sets of 3 mice from each of the remaining 5 groups were used to generate Figures 2 to 5 . At the end of the experimental period, the mice were killed by cervical dislocation after inhalation anesthesia with diethyl ether (Funakoshi, Tokyo, Japan), which was applied to cotton wool and placed with the mouse in an enclosed space. All animal procedures, including care and handling, conformed to the guidelines of our Institutional Animal Care and Use Committee, and the study protocol was approved by the Animal Research Committee of Showa University.
Three mice were killed at 21 days of age. The remaining mice were randomly divided into 5 groups of 9 when weaned: 18 mice were fed the SD, with 9 killed at 4 weeks of age and 9 killed at 7 weeks of age; 18 mice were fed the HD, with 9 killed at 4 weeks of age and 9 killed at 7 weeks of age; and 9 mice were fed the HSD every other week, and then killed at 7 weeks of age. The HD group received an ordinary laboratory diet for mice in a hard pellet form. The SD group received the ordinary diet after it was ground and mixed with water in standardized proportions (2 parts food to 5 parts water). No significant differences in weight were identified among the mice in the any of the groups either at randomization or when they were killed.
Tissue processing for hematoxylin and eosin and immunohistochemical staining were carried out. The dissected cartilage specimens from the mandibular condyles were fixed in 4% paraformaldehyde solution (pH 7.4) overnight at 4°C and then decalcified in 10% Na 2 EDTA (pH 7.2) for 2 weeks. Ten specimens from each group were embedded in paraffin and sectioned (7-μm thickness). The slices were coronal sections, which were carefully selected at the center of the mandibular condylar cartilage.
Five condylar sections from each group were stained with hematoxylin and eosin. The condylar cartilage was divided into 2 parts, comprising the articular, proliferative, and chondroblastic zones in one part, and the hypertrophic zone in another. The representative thicknesses were composed of the mean measurements of these zones.
Immune complexes were formed by mixing a mouse antibody against proliferating cell nuclear antigen (1:200 dilution; Oncogene, Boston, Mass) and EnVisionHRP antimouse detection reagent (Dako, Copenhagen, Denmark) for 1 hour at room temperature. After 60 minutes at room temperature, normal mouse serum (Dako code X0910) was added to the immune complex.
Five sections from each group were incubated with polymer immune complexes for 1 hour at room temperature. Deparaffinized paraffin sections were incubated with HRP-labeled primary antibodies using diaminobenzidine as the substrate. Equivalent dilutions and concentrations of mouse IgG2a (1:200 X0943; Dako) were used as negative controls.
Five sections from each group were incubated for 2.5 hours at room temperature with an antitype-X collagen antibody (1:100 dilution LSL-LB-0092 whole rabbit serum; LSL, Tokyo, Japan), which was detected using an Alexa Fluor 568-conjugated goat antirabbit polyclonal IgG (H+L) (Molecular Probes, Eugene, Ore) as a secondary antibody. The sections were observed with a fluorescence microscope (Eclipse TE300; Nikon, Tokyo, Japan) and an imaging system (AQUA COSMOS; Nikon).
Equivalent dilutions and concentrations of rabbit immunoglobulin fraction (1:100 X0903; Dako) were used as the negative controls.
We performed immunohistochemistry for osteopontin. Five sections from each group were incubated for 3 hours at room temperature with an antiosteopontin antibody (1:100 dilution LSL-LB-4225 whole rabbit serum; LSL), which was detected by incubation with EnVision + HRP antirabbit detection reagent (Dako) for 1 hour at room temperature. To prevent nonspecific staining, we incubated the sections with a blocking reagent containing normal mouse serum for an additional 1 hour at room temperature. Deparaffinized paraffin sections were reacted with HRP-labeled primary antibodies using diaminobenzidine as the substrate.
Equivalent dilutions and concentrations of rabbit immunoglobulin fraction (1:100 X0903; Dako) were used as negative controls.
Five sections from each group were incubated for 1 hour at room temperature with an anti-Musashi1 antibody (1:10 dilution 1877-1 rabbit Monoclonal IgG; Epitomics, Burlingame, Calif), which was detected by incubation with EnVision + HRP antirabbit detection reagent for 1 hour at room temperature. To prevent nonspecific staining, we incubated the sections with a blocking reagent containing normal mouse serum for an additional 1 hour at room temperature. Deparaffinized paraffin sections were reacted with HRP-labeled primary antibodies using diaminobenzidine as the substrate. In all experiments, negative controls were performed in which the primary antibody was omitted. Equivalent dilutions or concentrations of rabbit immunoglobulin fraction (1:10 X0903; Dako) were used as negative controls.
For the reverse transcription and real-time PCR, mandibular condylar cartilage was collected by dissection under a light microscope and snap-frozen in liquid nitrogen. Total RNA from 3 mice (nonpooled) was extracted from the total cartilage layer of the mandibular condyle and the tibial cartilage using an RNeasy Protect Mini Kit (Qiagen, Hilden, Germany) with RNase-free DNaseI treatment (Qiagen). Following the manufacturer’s protocol, we were able to extract sufficient quantities from 1 mouse. In addition, we confirmed by absorption spectrometer that the mRNA obtained was adequate and of good quality. First-strand cDNA was synthesized from 5 μg of total RNA using Super Script II RT (Gibco, Grand Island, NY) and random hexamers (Gibco). For real-time PCR, we used an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Darmstadt, Germany) with TaqMan probes. The final mixture (20 μL) contained 4 μL of cDNA sample, 10 μL of TaqMan Universal PCR Master Mix (Perkin-Elmer Applied Biosciences, Foster City, Calif), and 1 μL of primers for osteopontin, type X collagen, P75, Wnt-1, Musashi1, and Nestin (Mm00436767-m1Spp1, lot 455039; Mm00487041-m1, lot 479307; Mm00446296-m1, lot 958101; Mm01300555-g1, lot 812039; Mm00485224-m1, lot 649865; and Mm00450205-m1, lot 74463). Gene expression levels were normalized by the corresponding expression level of glyceraldehyde-3-phosphate dehydrogenase as a housekeeping gene. The PCR conditions were 2 minutes at 50°C, 10 minutes at 95°C, and 40 cycles of 15 seconds at 95°C and 1 minute at 60°C.
Statistical analysis
Measurements were performed on 3 separate tissue sections for each anatomic area in each experimental animal, and an average value was calculated. For gene expression, each sample of RNA was tested on 3 independent occasions, and the values were averaged. Averaged values from 3 representative mice from each dietary group were used for statistical analyses.
The significance of the differences between the independent samples was assessed using the Student t test (when comparing 2 groups) or 1-way analysis of variance (ANOVA) (when comparing 3) if the samples passed a preliminary Shapiro Wilks test for normality. Statistical significance was set at P <0.05. Because statistical tests were used mainly for exploratory analysis, multiple comparisons were not considered in this study. Our sample size of 3 animals for each test was not large enough to detect statistically significant differences between or among groups. However, since the variances of the data obtained within the groups were moderately small and well controlled, the results of the statistical tests provided useful information for our evaluation. All data are presented as means ± standard deviations in the figures. All analyses were performed using SPSS software (version 12.0; IBM, Chicago, Ill).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


