Introduction
In this study, we aimed to explore the expressions of α-smooth muscle actin, collagen type I, collagen type III, and osteocalcin in the periodontal ligament (PDL) under orthodontic loading, and to investigate the effect of myofibroblasts on collagen synthesis and osteocalcin production.
Methods
The teeth in the right maxillae of the rats were orthodontically loaded while the contralateral teeth remained unloaded as controls. The total 30 rats were divided into 5 groups, with each group corresponding to a treatment duration (0, 3, 5, 7, or 14 days, respectively). The expressions of α-smooth muscle actin, collagen type I, collagen type III, and osteocalcin in the tension area of the PDL over time were analyzed by immunochemistry staining. For the in-vitro study, the expressions of α-smooth muscle actin, collagen type I, collagen type III, and osteocalcin in the myofibroblasts and human osteoblast-like cells (MG63) coculture and PDL cells-MG63 coculture systems were examined by Western blot and real-time polymerase chain reaction.
Results
Enhanced expression of α-smooth muscle actin, collagen type I, collagen type III, and osteocalcin in the tension area of the PDL under orthodontic loading were observed in vivo, and increased expressions of α-smooth muscle actin, collagen type I, collagen type III, and osteocalcin in the myofibroblasts-MG63 coculture system were observed compared with the controls.
Conclusions
Expressions of α-smooth muscle actin, collagen type I, collagen type III, and osteocalcin are up-regulated in the PDL under orthodontic tensile loading. Myofibroblasts have a more positive effect on collagen synthesis and osteocalcin expression than do PDL cells.
Orthodontic tooth movement is the result of biologic responses to interference in the physiologic equilibrium of the tooth-alveolus complex by an externally applied force, during which mechanical-load transduction into bone response is of particular concern. The periodontal ligament (PDL) might be both the medium of force transfer and the means by which alveolar bone remodels in response to applied forces. The resulting stresses or strains within the PDL and the bone control the bone-modeling process. The typical fibril-forming collagen type I (Col-I) and collagen type III (Col-III) are the major collagen constituents of the extracellular matrix in the PDL, although Col-I especially predominates. Both collagen constituents of the extracellular matrix and resident cells constitute the structural basis for the biomechanical properties of the PDL. Thus, investigations of remodeling of the extracellular matrix and differentiation of the resident cells would help to clarify the biologic adaptation of the PDL to mechanical loading.
In our recent study, myofibroblasts were demonstrated to reside in the PDL under orthodontic loading and to have phenotypic features with the capacity to participate in orthodontic force transmission and periodontal tissue remodeling.
Myofibroblasts are mechanically active and are characterized by enhanced cellular contractile force generation due to the expression of α-smooth muscle actin (α-SMA)-containing stress fibers, which are also the myofibroblast phenotypic markers. Myofibroblast contractile force and resistant extracellular matrix render a positive feedback loop of increased intracellular contractile activity and extracellular stress. Myofibroblasts act as motors for tissue remodeling through the secretion of Col-I and Col-III and contribute to alterations in the extracellular mechanical microenvironment and overall tissue mechanics. This mechanosensitive cell has been extensively studied in the field of connective tissue remodeling, and sustained myofibroblast activation can drive pathologic conditions such as scarring and fibrosis.
To date, little is known about the significance of myofibroblast differentiation during orthodontic tooth movement. Both PDL cells and osteoblasts reside in the PDL. Osteoblasts line the alveolar bone surfaces close to the PDL cells. This close proximity inspired us to investigate the possible interactions between the 2 types of cells, especially when some PDL cells should largely be considered to be myofibroblasts under orthodontic loading. We extended this work to explore the expressions of α-SMA, Col-I, Col-III, and osteocalcin in the PDL under orthodontic loading, and to investigate the effect of myofibroblasts on collagen synthesis and osteocalcin production, with α-SMA used as the functional marker for myofibroblasts and osteocalcin as the osteogenic marker of the later stages of osteoblast differentiation and bone formation.
However, α-SMA and osteocalcin would not act as phenotypic markers for cell types when the cells are under stimulation. It was reported that cells with a pericyte-myofibroblast phenotype have the potential to differentiate into functional osteoblasts and that osteoblasts can display contractile behavior through expression of α-SMA. It becomes a requisite to conduct in-vitro experiments, which allow more reliable and specific identification of myofibroblasts by coexpression of vimentin and α-SMA without expression of desmin.
Mechanical strain in combination with transforming growth factor (TGF)-β is recognized as a prerequisite for myofibroblast differentiation, with TGF-β1 as the most potent inducer. In this study, we conducted cell cultures in vitro for myofibroblast identification, using mechanical strain application or TGF-β1 treatment as the induction condition. To explore the effect of myofibroblasts on collagen and osteocalcin production, we focused on specific cell types using myofibroblast-MG63 coculture model in vitro.
Human osteoblast-like cell MG63 is a well-characterized cell line exhibiting many osteoblastic traits characteristic of bone-forming cells and has frequently been used in vitro for determining cell differentiation and cell activity. In establishing the myofibroblast-MG63 coculture system, the PDL cells were treated with TGF-β1 first and then cocultured with MG63 cells after removal of the TGF-β1. Thus, stability of TGF-β1–induced myofibroblastic phenotype was examined preliminarily.
Cellular fibronectin, synthesized by fibroblasts and osteoblasts, plays a fundamental role in cell-to-cell and cell-to-extracellular matrix interactions. Cell adhesion to fibronectin, mediated by integrin transmembrane receptors, initiates intracellular signaling cascades that regulate morphology, proliferation, and differentiation of cells, including osteoblasts and fibroblasts. Hence, we focused on fibronectin to explore the mechanism by which myofibroblasts and MG63 cells might interact.
Material and methods
A total of 30 male Sprague Dawley rats (age, 8 weeks; weight, 200 ± 20 g) were divided into 5 groups. Animal interventions were approved by the institutional committee for animal care of Sichuan University, Chengdu, China. An animal model for orthodontic tooth movement was established as described in a previous study. Briefly, after the rats were anesthetized, continuous orthodontic force was supplied by a closed-coil spring (3M Unitek, Monrovia, Calif) with one end fixed to the maxillary incisor and the other end to the maxillary first molar. Teeth in the right maxillae were ligated to the appliance, with the spring activated to exert an initial force of 40 g with no further reactivation during the experiment; the contralateral teeth served as the controls. The total 30 rats were divided into 5 groups, with each group corresponding to a treatment duration (0, 3, 5, 7, or 14 days, respectively). After the rats were killed by an overdose of anesthesia, the maxillae were removed and fixed in 4% paraformaldehyde at room temperature for 24 hours. Decalcification of tissues was performed using 10% ethylenediaminetetraacetic acid for 3 weeks. The demineralized tissues were dehydrated and embedded in paraffin and then cut into 6-μm-thick mesiodistal serial sections for immunohistochemistry staining.
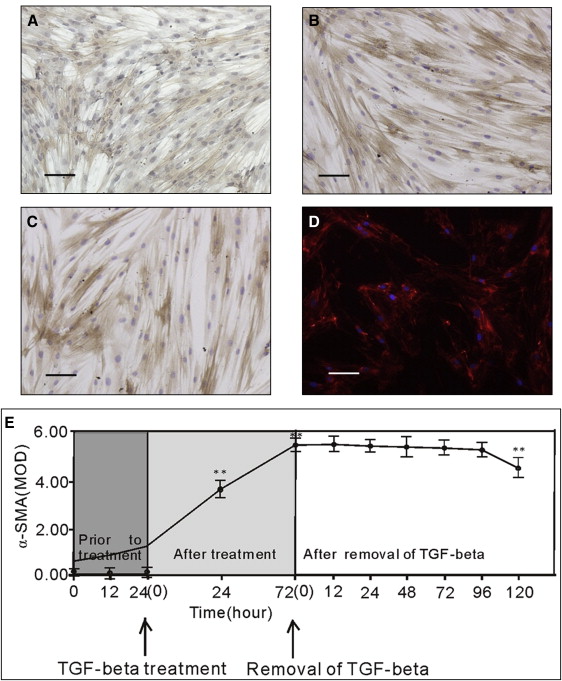
The preparation of human PDL cells was performed according to the method previously described by Palmon et al. Briefly, the cells were isolated from healthy PDLs of premolars of teenagers undergoing tooth extractions for orthodontic reasons. All patients gave informed consent before providing the samples. The periodontal tissues were removed from the central part of the root surface. The cells from the explants were cultured for 7 days and then passaged at a 1:3 split ratio after the cells had reached confiuence. Cells between the third and fifth passages were seeded at a density of 1 × 10 5 cells per square centimeter in 6-well plates and cultured in Dulbecco’s modified eagle medium (DMEM) plus 10% fetal bovine serum (FBS). After reaching confluence, cells were made quiescent by incubation with serum-free medium for 24 hours. The serum-free medium was then removed and replaced by DMEM plus 0.1% FBS. For vehicle control, the cells were maintained in DMEM plus 0.1% FBS for 72 hours; for stimulating differentiation of myofibroblasts, human recombinant TGF-β1 (5 ng/mL; Protech Technology, Sparks, Nev) was added to the DMEM and 0.1% FBS medium, and cells were maintained in the culture for 72 hours, or the cells were subjected to cyclic uniaxial tensile stress (magnitude, 4000 μstrains; frequency, 0.5 Hz) for 6 hours. The stress was supplied by a uniaxial 4-point bending system (developed at Sichuan University; patents CN2534576 and CN1425905). After 72 hours, the TGF-β1–containing medium was changed to DMEM and 0.1% FBS, and cells were harvested at 0, 12, 24, 48, 72, 96, and 120 hours after removal from the TGF-β1. Cells were prepared for α-SMA staining by immunochemistry and immunofiuorescence. Human osteoblast-like cells (MG63) were thawed from a frozen stock, cultured, and passaged in DMEM and 0.5% FBS at 37°C with 5% carbon dioxide. Early passage cells were used for cocultures.
Myofibroblast-MG63 coculture and PDL cell-MG63 coculture models were established to compare the effect of myofibroblast (induced by TGF-β1 in vitro) on collagen and osteocalcin production with that of the PDL cells. First, human PDL cells were seeded in 6-well plates and incubated in DMEM and 10% FBS for 24 hours at 37°C under 5% carbons dioxide, at a density of 4 × 10 4 per square centimeter for immunochemistry staining and 4 × 10 5 per square centimeter for Western blot and real-time polymerase chain reaction (RT-PCR) analysis. The cells were then made quiescent by incubation in serum-free medium for 24 hours. Second, the serum-deprived human PDL cells were cultured in DMEM and 0.1% FBS with 5 ng per milliliter of TGF-β1 treatment, or in DMEM and 0.1% FBS of identical quantity without TGF-β1 treatment (vehicle control), for 72 hours. Last, for the myofibroblast-MG63 coculture, the medium with TGF-β1 treatment was changed to DMEM and 0.5% FBS containing MG63 cells (2 × 10 4 per square centimeter for immunochemistry staining and 2 × 10 5 per square centimeter for Western blot and RT-PCR analysis); for the PDL cells-MG63 coculture (vehicle control), the DMEM and 0.1% FBS without TGF-β1 treatment was changed to DMEM and 0.5% FBS containing MG63 cells (also 2 × 10 4 per square centimeter or 2 × 10 5 per square centimeter). Thereafter, both myofibroblast-MG63 cocultures and PDL cells-MG63 cocultures were divided into 5 groups, with each group maintained for a different duration (0, 12, 24, 48, or 72 hours, respectively). Coculture cells harvested at each time point were prepared for assays: expressions of α-SMA, osteocalcin, and fibronectin were visualized by immunochemistry staining; α-SMA, Col-I, Col-III, and osteocalcin expression levels were examined by RT-PCR and Western blot.
Immunochemistry staining was performed as described previously using monoclonal antibodies to α-SMA (1:50 dilution, CB4 0FL; Abcam, Cambridge, United Kingdom), monoclonal antibodies to Col-I (1:200; Abcam), polyclonal antibodies to Col-III (1:100; Abcam), and monoclonal antibodies to osteocalcin (1:100; Santa Cruz Biotechnology, Santa Cruz, Calif). Monoclonal mouse anticytokeratin (1:100; Dako, Glostrup, Denmark), monoclonal mouse antivimentin (1:100; Santa Cruz Biotechnology), monoclonal antidesmin (1:100; Dako), and monoclonal mouse antifibronectin (1:100; Santa Cruz Biotechnology) were used as the primary antibodies. Biotinylated horse antimouse immunoglobulin G and goat antirabbit immunoglobulin G (1:100; Zhongshan Biotechnology, Beijing, China) were used as the secondary antibodies. The reaction products were visualized in brown with a diaminobenzidine substrate kit (DAB; Zhongshan Biotechnology) or in persicine with alkaline phosphatase immunostaining (AP-Red; Zhongshan Biotechnology).
Manually selected study areas were viewed with a microscope (E600; Nikon, Tokyo, Japan). The study areas were selected in the PDL distal to the cervical third of the roots of the maxillary first molars, at a constant distance from the root surfaces. These selected areas were definitely tension regions no matter what pattern of tooth movement (tilting or bodily) was created. The size of the study area and the integrated optical density of the positive stains were measured by Image-Pro Plus (5.0; Media Cybernetics, Bethesda, Md). Protein expressions were measured by the mean optical density (mean optical density = integrated optical density per unit of study area). Each sample was analyzed in triplicate.
Immunofiuorescence staining was performed as described previously using monoclonal antibodies to α-SMA (1:100; Abcam), and 4′,6-diamidino-2-phenylindole (DAPI; 1:500; Invitrogen, Paisley, United Kingdom) was used to demonstrate nuclear localization. Subsequently, cells embedded in 50% glycerol were mounted and observed under a fluorescence microscope (Bio-Rad, Hercules, Calif).
RT-PCR analysis was performed according to the manufacturer’s instructions. Total RNA was extracted from the cocultured cells using Trizol (Invitrogen, Carlsbad, Calif). The RNA samples used in the analysis were high quality (with the A260/280 ratios from 1.92-1.99, and the RNA Integrity Number values from 7.14-9.65). cDNA was reverse transcribed from 2 μL of total cellular RNA using an RT-PCR kit (Takara, Tokyo, Japan). We subjected 5 μL of the cDNA mixture to PCR amplification. Oligonucleotide primers and TaqMan probes for α-SMA, Col-I, Col-III, and osteocalcin were designed from the GenBank data bases. The primer sets were 5′-AAGATGACCCAGATCATGTT-3′ (forward) and 5′-CACAATGTCCCCATCTATGA-3′ (reverse) for α-SMA, 5′-GTGCTAAGGGTGAAGCTGGT-3′ (forward) and 5′-CATCAGCACCAGGGTTTCCAG-3′ (reverse) for Col-I, 5′- CAGGGGCCCCAGGACTTAGAG-3′ (forward) and 5′-GGGCCAGGAGGACCAATAGGA-3′ (reverse) for Col-III, and 5′-GTGCAGCCTTTGTGTCCAAG-3′ (forward) and 5′-GTCAGCCAACTCGTCACAGT-3′ (reverse) for osteocalcin. Target genes were all normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
For the Western blot analysis, total cellular protein was extracted in a lysis buffer and separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a polyvinylidene fluoride membrane (Roche, Grenzach-Wyhlen, Germany). Membranes were blocked with 5% bovine serum albumin for 1 hour at room temperature and subsequently incubated overnight at 4°C with the primary antibodies of α-SMA, Col-I, Col-III, osteocalcin, and GAPDH (all from Abcam), respectively, as well as the horseradish peroxidase-conjugated secondary antibodies (1:5000 dilution; Millipore, Billerica, Mass). The immunoreactive bands were visualized by enhanced chemiluminescence (Santa Cruz Biotechnology) and analyzed by Quantity One 1-D software (Bio-Rad); all protein expression was normalized to GAPDH.
Statistical analysis
Statistical analyses were performed by 1-way analysis of variance. Least significant difference test and Student-Newman-Keuls test were used for multiple comparisons between pairs. Statistical significance of the correlations was determined using the Pearson product-moment method; P <0.05 was considered to be statistically significant. All data are presented as the means and standard deviations of the triplicate determinations. Each experiment was repeated 3 times with similar results.
Results
The effect of orthodontic loading on the expressions of α-SMA, Col-I, Col-III, and osteocalcin in the tension area of the PDL over time was semiquantitively analyzed by immunochemistry staining and image analysis ( Fig 1 ). Time course analysis showed that α-SMA production increased progressively from 0 to 14 days and reached a maximum at day 14. There were time-dependent, statistically significant increases in the production of Col-I and Col-III throughout the experiment, except for a decline in Col-III production from 7 to 14 days. The increase in osteocalcin production was mild initially and significant later, and the time-dependent increase in osteocalcin production seemed to lag behind the change in α-SMA production.
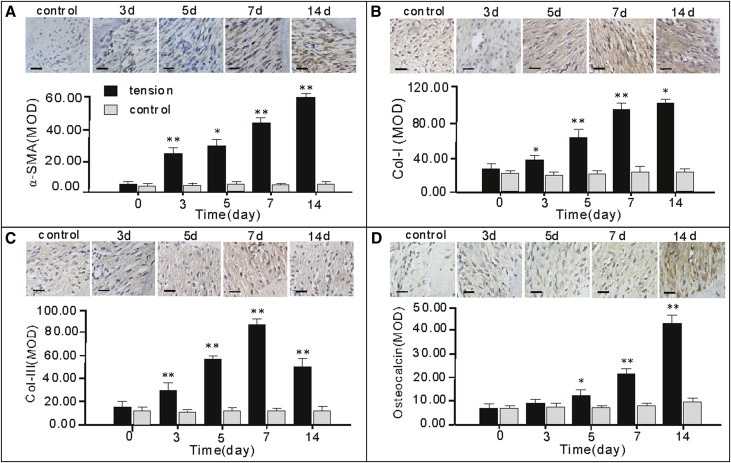
Statistical analysis of the mean optical density values for the 4 types of proteins showed a highly correlated relationship of α-SMA with Col-I, Col-III, and osteocalcin expression. There were high correlations between the expression levels of α-SMA and Col-I (n = 13, r = 0.927, P <0.001; data not shown), between the expression levels of α-SMA and Col-III (n =13, r = 0.945, P <0.001; data not shown), and between the expression levels of α-SMA and osteocalcin (n =13, r = 0.891, P <0.001; data not shown).
A relatively stable myofibroblastic phenotype was induced by TGF-β1 in vitro until 72 hours after its removal. Stimulation of PDL cells with 5 ng per milliliter of TGF-β1 or 4000 μstrains of tensile stress resulted in a phenotypic alteration characterized by increased expression of α-SMA ( Fig 2 ), and 5 ng per milliliter of TGF-β1 treatment induced definite differentiation from PDL cells to myofibroblasts, identified by coexpression of vimentin and α-SMA without expression of desmin and cytokeratin ( Fig 3 ). Treatment of human PDL cells with TGF-β1 for 72 hours and then removal of TGF-β1 did not lead to an immediate reversal of the myofibroblastic phenotype. After removal of TGF-β1, flow cytometry performed on the cells showed an almost unchanged ( P >0.05) ratio of cells at G0 phase to G1 phase until 72 hours (indicating unchanged proliferation rate), although with a statistically significant difference at 96 hours ( P <0.001; data not shown). Time-course analysis of α-SMA production showed no statistically significant ( P >0.05) difference until 96 hours ( Fig 2 ), suggesting a relatively stable myofibroblastic phenotype induced by TGF-β1 until 72 hours after its removal. Based on the findings about the time limit of stability of TGF-β1–induced myofibroblastic phenotype, the myofibroblast-MG63 coculture model was established with an observation time of 72 hours.