This article addresses several issues in the approach to diagnosis of oral cancer. The term oral cancer is clarified. Key aspects of the biologic basis of development of oral cancer and the known risk factors associated with the disease are summarized. The clinical presentation of oral cancers and precancerous lesions and their histopathologic correlates is discussed. The importance of conventional tissue biopsy as the prevailing gold standard for diagnosis is emphasized. Other current technologies available for detecting and diagnosing oral cancer and premalignant lesions are acknowledged, and their respective strengths and weaknesses are discussed.
Oral cancer is a diagnosis that patients fear and every oral health care provider dreads having to convey. In daily practice of dentistry and medicine, most pathologic conditions are reactive, inflammatory, and perhaps infectious in nature. Malignant neoplasms, including oral cancer (ie, squamous cell carcinoma [SCC]), occur infrequently. Why then, is the fear of oral cancer so pervasive and intense? Almost certainly, it is because both the disease and its treatment conjure a unique spectrum of images associated with profound morbidity. Mental portraits of disfigurement, unrelenting function loss, protracted pain, and even death decrease within the cascade of associations triggered by a diagnosis of oral cancer. Risk for recurrent disease, distant metastases, and susceptibility for developing second primary carcinomas of the upper aerodigestive tract as a product of field cancerization are among the biologic specters linked to a diagnosis of oral cancer.
Despite important advances in the approach to treatment of oral cancer, 5-year survival from the time of diagnosis has remained disappointingly static over the past 50 years. What accounts for this dismal prognosis? The answer to that question is multifaceted, but the poor outlook is in part attributable to late-stage diagnoses with advanced disease. In more than 50% of cases of oral cancer, tumors have already spread distantly before being diagnosed. This situation suggests that many providers, or their patients, or both, are either failing to recognize early oral mucosal changes that indicate cancer development or they are evading timely clinical evaluation of such findings. In stark contrast, in cases in which a primary tumor is localized and there is no evidence of metastasis to locoregional lymph nodes at diagnosis, the outlook for survival at 5 years is significantly better. What does this tell us? That the earlier an oral cancer is diagnosed, the better the prospects for improved survival. Given the ready anatomic accessibility of the oral cavity for visual and tactile examination, detecting potentially malignant (ie, precancerous, preinvasive) oral mucosal lesions before they have attained even incipient malignant status should not be insurmountable.
How can clinicians best position themselves to diagnose oral cancer at an early clinical stage of the disease? They must be informed. Contemporary clinicians must have an understanding of the pathogenesis of a disease in which oral surface epithelium that is presumably normal at outset transforms to a malignant state over an extended period. Such understanding must include awareness of the environmental influences, internal and external, that are associated with this transformation, and that are instrumental for identifying individuals at greatest risk for developing the disease. Informed clinicians are best prepared to recognize the spectrum of clinical findings that are most suspicious for evolving (ie, premalignant) or incipient oral cancer lesions. They know the most appropriate approach to take when a suspect oral lesion has been discovered, because that ensures that an accurate diagnosis is obtained expeditiously. They are aware of the long-term implications of a diagnosis of oral precancer or cancer, so that a reasonable and realistic plan for proper clinical follow-up is instituted.
Several issues are addressed in our discussion of contemporary concepts in the approach to diagnosis of oral cancer. We should clarify the term oral cancer. The literature is replete with statistics concerning oral cancer referable to a single broadly diverse anatomic region that encompasses the lips, the oral cavity proper, and the tonsils, oropharynx, and larynx. The problem with combining these various sites into a single location is that there is mounting evidence to suggest that the SCCs that arise in each of these respective regions are associated with different risk factors, morbidities, and progressions toward mortality. Therefore, much of our discussion of oral cancer epidemiology and diagnosis is confined to SCCs of the oral cavity proper, that is, the anatomic region that extends from the mucosa of the lips anteriorly, to the anterior tonsillar pillars and the oral portion of the tongue, posteriorly. Key aspects of the biologic basis of oral cancer development, and the known risk factors associated with the disease are summarized. The clinical presentation of oral cancers and precancerous lesions, and their histopathologic correlates, is discussed. The importance of conventional tissue biopsy as the prevailing gold standard for diagnosis is emphasized. Other current technologies available for detecting and diagnosing oral cancer and premalignant lesions are acknowledged, and their respective strengths and weaknesses are discussed.
Epidemiology
Of 1.5 million cancers diagnosed in the United States annually, oral cancer (SCC) accounts for less than 3% of all cases. Globally, it is the sixth most common cancer. The incidence is especially high in the Indian subcontinent, Brazil, Sub-Saharan Africa, Australia and other regions. Oral SCC is the most common malignancy in Southeast Asia.
Incidence of Oral SCC
In a review of statistics, the SEER (Surveillance Epidemiology and End Results) data estimated that 35,720 individuals (25,240 men and 10,480 women) would be diagnosed with cancer of the oral cavity and pharynx in 2009, and that 7600 men and women in the United States would die of the disease. Reading these estimates, the epidemiologic factors and pathogenetic mechanisms that contribute to oral cavity, pharyngeal, and laryngeal SCCs, respectively are similar, but not identical. However, cancer-related data from these sites are often presented collectively. This strategy has resulted in the mistaken implication that the data pertain to a single disease entity. Similarly, several studies also include data from lip cancers in their discussions of oral cancer. However, the cause of cancer of the external lip differs considerably from that of cancer of the oral cavity, and the pharynx and larynx. Therefore, lip cancer data should be considered separately.
It is important to define the anatomic boundaries of the oral cavity to better understand the unique incidence, pathogenetic factors, and consequences of oral SCC. The oral cavity begins anteriorly at the vermilion-mucosal junction of the lips and extends posteriorly to include the base of the oral tongue and the anterior tonsillar pillars. It does not include the pharynx and larynx. Separated in this manner, SCCs of the oral cavity proper (ie, excluding the tonsils, pharynx, larynx, and vermilion-skin portions of the lips) account for approximately 17,000 new cancer cases annually and result in an estimated 6000 deaths per year.
Age, Race, and Sex
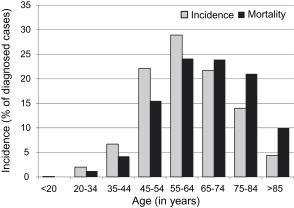
It has been long recognized that oral SCC, like many other neoplasms, is a disease of age ( Fig. 1 ). In 2009, the median age at diagnosis for SCC of the oral cavity was 61 years. The increased incidence of oral SCCs with age is consistent with the current understanding that, like most malignant neoplasms, oral cancer results from mutations accumulated over a long period. These mutations result in irreversible multifaceted deregulation of cellular homeostasis and eventuate in aberrant differentiation, growth, and replication that, in turn, affect genetic stability, cell aging, apoptosis, carcinogen-mediated mutagenesis, and immune surveillance.
In recent decades the incidence rate for oral SCC in men has steadily declined, whereas in women it has remained stable. This finding translates into a relative increase in incidence of oral SCC in women. From 1975 to 2006, both black and white women had similar incidence rates of oral SCC; however, black men have consistently had a 30% higher rate than white men. Similar findings have been reported relative to associated mortality in African American men with oral SCC: the rates remained generally stable or increased between 1975 and 2006. Asian and Hispanic people manifest the lowest rates of oral SCC in the United States.
Site-specific Epidemiology
In the United States, SCC of the oral tongue is the most commonly reported site (36.2% of all oral SCC), followed closely by the floor of the mouth (24.5%). As discussed in the section on clinical features, the lateral tongue and the floor of the mouth comprise more than 60% of the so-called cancer-prone locations in the oral cavity. Other locations that figure prominently in the SEER data are the tonsillar pillars and retromolar pad areas (20%), followed by the soft palate, lower labial mucosa, buccal mucosa, and gingiva (∼19%). In the United States the buccal mucosa is the sixth most common oral cancer site. Only gingival carcinomas are less common. In striking contrast, the buccal mucosa is the most common location for oral SCC in some other parts of the world (Central and Southeast Asia). The differences in site-specific incidence are likely attributable to cultural differences in the way tobacco and related products are used relative to their mode of delivery (smoking vs chewing). Direct mucosal contact with smokeless tobacco and other noncombustible preparations (eg, paan/betel nut/gutka) seems to account for the predominance of buccal and gingival carcinomas in the non-US oral SCC cases mentioned earlier.
Stage at Diagnosis and Survival
The clinical staging of any cancer defines the extent of disease in terms of anatomic spread, and is vital to the development of a treatment plan. It also provides the framework that permits comparison of treatment strategies, outcomes, and survival rates. The universally accepted TNM (Tumor, Node, Metastasis) staging system is used worldwide in staging malignant processes. Staging is site-specific and ranges from stage I (local disease) to stage IV (disseminated disease).
Of oral SCCs, 46% are diagnosed after the cancer has spread to locoregional lymph nodes (stage III); 35% have localized disease (stage I or II); 13% present with distantly disseminated metastatic disease (stage IV); and 6% present with unknown stage. The corresponding 5-year survival rates are 55.1% (stage III), 77.2% (stage I and II), and 29.1% for disseminated disease (stage IV). This finding means that approximately 60% of oral SCCs are diagnosed at stage III or IV with a correspondingly low 5-year survival rate.
Statistics and detailed numbers notwithstanding, these data highlight 2 important facts:
- A.
Advances in treatment in the past several decades have not led to significant improvement in survival
- B.
Early diagnosis is a key factor in improving survival rate, thereby improving the morbidity and mortality associated with oral SCC.
Therefore, it is vital that dentists and hygienists, who serve as the gatekeepers of the oral cavity, understand the disease and recognize the clinical features of oral SCC and oral precancerous lesions.
Cause and pathogenesis
Oral SCC is a malignant neoplasm that arises from the squamous epithelium that lines the oral cavity. Familiarity with the general structure and function of squamous epithelium is essential for understanding the etiopathogenesis of oral SCC. The following review of the general pathogenetic mechanisms that drive malignant neoplastic progression provides the framework on which the various cause/risk factors of oral SCC are discussed.
Stratified squamous epithelium (unkeratinized/keratinized) lines the entire oral mucosal surface. Similar to any other type of epithelium, it rests on a complex fishnetlike basement membrane to which it is tethered by hemidesmosomes. Keratinocytes, the cells that serve as the functional units of all squamous epithelia, together form a meshwork of interconnected cells that comprise stratified squamous epithelium. The primary role of a keratinocyte is to proliferate and ascend from its origin in the basal-most stratum (basal cell layer) to the epithelial surface. As they ascend through the epithelial strata, keratinocytes are genetically programmed to differentiate, mature, undergo natural death, and finally leave a protective protein product, keratin, on the epithelial surface. Local homeostasis, especially in the basal and suprabasal epithelial layers, is tightly regulated by a series of genes that play critical roles in controlling the cell cycle of the keratinocyte ( Table 1 ). Progression through the cell cycle is dependent on a combination of growth factors, growth suppressors, telomere length, and nutrition (vascular supply), and is regulated by checkpoint genes (eg, p53, p21, Rb), programmed cell death (apoptosis), local immune surveillance, and other factors. In addition, the components of the basement membrane (the structure that provides a natural barrier between the epithelium and the underlying superficial submucosa [lamina propria] and serves as the foundation for the overlying epithelial strata) are maintained in a state of equilibrium by protease and antiprotease activity. Any lasting change (eg, acquired genetic defects, dysregulated immune function) that disturbs the local equilibrium potentially results in uncontrolled keratinocyte proliferation and/or loss of integrity of the epithelium-basement membrane relationship.
| Pathogenetic Mechanisms | GOF/LOF Mutations |
|---|---|
| A. Self-sufficiency in growth signals | A. Autocrine signaling (eg, GOF of EGF, TGFβ, KGF) |
| B. Insensitivity to antigrowth signals | B. LOF of tumor suppressors (eg, p16, p53, p21, CDK4/6) |
| C. Acquired capability to evade apoptosis | C. GOF of antiapoptotic genes or LOF of proapoptotic genes |
| D. Limitless replicative potential | D. Telomere lengthening (GOF of telomerase) |
| E. Sustained angiogenesis, stromal support | E. GOF mutations of angiogenic molecules (eg, VEGF, bFGF) |
| F. Tissue invasion and metastasis: Be independent of cell-cell interactions (loosen up) Acquire the ability to degrade the basement membrane and surrounding extracellular matrix Migration and locomotion Vascular/lymphatic dissemination and homing of tumor cells |
F. Tissue invasion and metastasis: Mutations/ altered function of E-cadherins, β-catenin, keratins, actin cytoskeleton GOF produces proteolytic enzymes (eg, collagenases, MMPs, cathepsin D) and LOF of proteolytic inhibitors (eg, TIMPs) Abnormal proteolytic cleavage/folding of extracellular matrix proteins (eg, integrins, laminins, tenascin) Acquire the ability to survive in an unattached state Avoid local immune surveillance (decoy surface molecules) Migrate and home to vascular channels (chemokinesis) Survive mechanical shear (anoikis) |
Normally, a range of inherent host regulatory/preventive mechanisms maintain orderly differentiation, growth, and integrity of the surface epithelium, so that to become a successful cancer cell an epithelial cell has to acquire certain properties that distinguish it from its surrounding normal counterparts. These distinguishing properties are acquired through mutations (see Table 1 ).
A cell must acquire several mutations to be a successful cancer cell. Studies show that oral SCC, like most malignant tumors, results from a protracted sequence of events that recur repeatedly over many years. This finding is consistent with epidemiologic data that oral SCC is a disease that affects adults primarily. Several cancer progression models that focus on a few commonly affected molecules and genes have been proposed. Nonetheless, the precise temporal sequence of mutations is different for each organ and tumor type, and varies from individual to individual.
Cause and pathogenesis
Oral SCC is a malignant neoplasm that arises from the squamous epithelium that lines the oral cavity. Familiarity with the general structure and function of squamous epithelium is essential for understanding the etiopathogenesis of oral SCC. The following review of the general pathogenetic mechanisms that drive malignant neoplastic progression provides the framework on which the various cause/risk factors of oral SCC are discussed.
Stratified squamous epithelium (unkeratinized/keratinized) lines the entire oral mucosal surface. Similar to any other type of epithelium, it rests on a complex fishnetlike basement membrane to which it is tethered by hemidesmosomes. Keratinocytes, the cells that serve as the functional units of all squamous epithelia, together form a meshwork of interconnected cells that comprise stratified squamous epithelium. The primary role of a keratinocyte is to proliferate and ascend from its origin in the basal-most stratum (basal cell layer) to the epithelial surface. As they ascend through the epithelial strata, keratinocytes are genetically programmed to differentiate, mature, undergo natural death, and finally leave a protective protein product, keratin, on the epithelial surface. Local homeostasis, especially in the basal and suprabasal epithelial layers, is tightly regulated by a series of genes that play critical roles in controlling the cell cycle of the keratinocyte ( Table 1 ). Progression through the cell cycle is dependent on a combination of growth factors, growth suppressors, telomere length, and nutrition (vascular supply), and is regulated by checkpoint genes (eg, p53, p21, Rb), programmed cell death (apoptosis), local immune surveillance, and other factors. In addition, the components of the basement membrane (the structure that provides a natural barrier between the epithelium and the underlying superficial submucosa [lamina propria] and serves as the foundation for the overlying epithelial strata) are maintained in a state of equilibrium by protease and antiprotease activity. Any lasting change (eg, acquired genetic defects, dysregulated immune function) that disturbs the local equilibrium potentially results in uncontrolled keratinocyte proliferation and/or loss of integrity of the epithelium-basement membrane relationship.
| Pathogenetic Mechanisms | GOF/LOF Mutations |
|---|---|
| A. Self-sufficiency in growth signals | A. Autocrine signaling (eg, GOF of EGF, TGFβ, KGF) |
| B. Insensitivity to antigrowth signals | B. LOF of tumor suppressors (eg, p16, p53, p21, CDK4/6) |
| C. Acquired capability to evade apoptosis | C. GOF of antiapoptotic genes or LOF of proapoptotic genes |
| D. Limitless replicative potential | D. Telomere lengthening (GOF of telomerase) |
| E. Sustained angiogenesis, stromal support | E. GOF mutations of angiogenic molecules (eg, VEGF, bFGF) |
| F. Tissue invasion and metastasis: Be independent of cell-cell interactions (loosen up) Acquire the ability to degrade the basement membrane and surrounding extracellular matrix Migration and locomotion Vascular/lymphatic dissemination and homing of tumor cells |
F. Tissue invasion and metastasis: Mutations/ altered function of E-cadherins, β-catenin, keratins, actin cytoskeleton GOF produces proteolytic enzymes (eg, collagenases, MMPs, cathepsin D) and LOF of proteolytic inhibitors (eg, TIMPs) Abnormal proteolytic cleavage/folding of extracellular matrix proteins (eg, integrins, laminins, tenascin) Acquire the ability to survive in an unattached state Avoid local immune surveillance (decoy surface molecules) Migrate and home to vascular channels (chemokinesis) Survive mechanical shear (anoikis) |
Normally, a range of inherent host regulatory/preventive mechanisms maintain orderly differentiation, growth, and integrity of the surface epithelium, so that to become a successful cancer cell an epithelial cell has to acquire certain properties that distinguish it from its surrounding normal counterparts. These distinguishing properties are acquired through mutations (see Table 1 ).
A cell must acquire several mutations to be a successful cancer cell. Studies show that oral SCC, like most malignant tumors, results from a protracted sequence of events that recur repeatedly over many years. This finding is consistent with epidemiologic data that oral SCC is a disease that affects adults primarily. Several cancer progression models that focus on a few commonly affected molecules and genes have been proposed. Nonetheless, the precise temporal sequence of mutations is different for each organ and tumor type, and varies from individual to individual.
Risk factors
The genetic mutations leading to oral carcinogenesis may be spontaneous in some individuals. However, most are acquired, and are associated with predisposing or etiologic risk factors. Classic carcinogenesis experiments show the importance of risk factors and break them down into initiators and promoters that either inaugurate or advance permanent DNA damage. A single risk factor frequently can play both roles. Some of the risk factors/mutagens associated with oral SCC are discussed next.
Tobacco
Tobacco, the most common cause of human cancers, is responsible for ∼85% of oral SCCs and ∼90% of lung cancers. Worldwide, cigarette smoking causes more than 5 million deaths annually. Numerous independent investigations in the past several decades have confirmed a link between oral SCC and tobacco smoking, including numerous case-control and cohort studies. The proportion of smokers (∼85%) among patients with oral SCC is nearly 3 times greater than the general population. In addition, studies show that the risk for a second primary carcinoma of the oral cavity/aerodigestive tract is ∼2 to 6 times greater in patients with a history of smoking. The number of potentially noxious/carcinogenic chemicals in tobacco smoke is extraordinary: it contains between 2000 and 4000 substances, 60 of which are identified carcinogens (nicotine, an alkaloid, is not a carcinogen but has been shown to be a potent addictive agent). Some substances generated by tobacco smoke that are recognized carcinogens include benzo[a]pyrene , TSNAs (tobacco-specific nitrosamines), phenol, and free radicals. Furthermore, studies consistently show a dose-response effect, with increased incidence of oral SCC related to the duration and frequency of smoking. There is about a 20-fold risk of oral SCC in heavier tobacco smokers. Although increased smoking is related to increased risk for oral SCC, the risk for developing oral SCC does not necessarily diminish after cessation of smoking.
As discussed earlier, the striking variations in oral SCC sites and incidence seen among different regional, cultural, and demographic groups are largely attributable to differences in the use and mode of delivery of tobacco. In the United States and much of the developed world cigarette smoking has been shown to have a linear dose-response carcinogenic effect. The risk for developing oral SCC remains the same, if not higher, in pipe and cigar smokers. Smoking tobacco products like hookahs (water pipes), bidis (hand-rolled, filterless tobacco cigarettes), and chutta (reverse smoking) are associated with a similarly high risk for oral SCC. Carcinogens from smoking that are released into the saliva tend to pool in the low-lying areas of the mouth and could account for the frequent occurrence of oral SCC along the lateral-ventral tongue and floor of the mouth. In Southeast Asia and the Middle East, where habitual use of other carcinogenic substances is common, the oral cancer-prone sites are different from those cited earlier. These patients’ tumors tend to occur more often in the vestibular, gingival, and buccal mucosae, as a result of placement of noncombustible carcinogenic substances in direct and prolonged contact with these areas.
Smokeless tobacco use and its association with oral SCC has been the subject of several studies. Studies from Scandinavia have found that the use of Swedish snuff did not increase the risk of oral SCC. However, some studies from the United States and around the world tell a different story. With the general upswing in snuff dipping and tobacco chewing, especially in the Southeastern United States, the incidence of oral SCC is higher than expected, especially in women. A case-control study of 255 women in North Carolina showed a 50-fold increased risk for SCC of the gingiva and buccal mucosa in long-term snuff dippers. The influence of smokeless tobacco on carcinogenesis seems to be associated with long-term use. Although studies often present conflicting opinions about smokeless tobacco, different risks are associated with different brands and products. These differences are attributable to the presence/absence of additives, flavoring agents, and modifiers that enhance the carcinogenic potential of the smokeless tobacco. Most tobacco products consumed in Asia and Africa are unregulated and are often of the smokeless variety. Products such as paan (mixture of betel leaf, lime, tobacco, catechu, and areca nut), gutka (tobacco, areca nut), mishri (powdered tobacco that is rubbed on the gums), and chutta (clumps of tobacco that are either chewed or reverse smoked) are strong risk factors for oral SCC.
Tobacco smoking (past or present) remains the most consistent risk factor for oral SCC in the United States. Yet, with the increased use of smokeless tobacco, especially in women and young individuals and athletes, increased awareness of the potential risks of those products and association with oral SCC is a vital data point in screening for the disease. Any history of tobacco use (past or present; smoking/ smokeless) must be viewed as a potential risk factor for oral SCC.
Alcohol
Alcohol is a well-recognized risk factor for several malignant neoplastic diseases, including oral SCC. It is recognized as a potent promoter of carcinogenesis. Ethanol is oxidized to acetaldehyde by alcohol dehydrogenase, releasing multiple free radicals. Although investigations that attempt to implicate alcohol alone as the main causative agent of oral SCC have yielded largely conflicting results, the correlation of alcohol with increased risk for oral SCC is indisputable. The major clinical significance of alcohol consumption seems to be its ability to potentiate the carcinogenic effect of tobacco. The effect is at least additive and may be multiplicative in individuals with heavy alcohol consumption or at sites with the highest levels of alcohol exposure. Although the underlying mechanisms for this association are poorly understood, some proposed mechanisms include:
- A.
Dehydration effects of alcohol render the mucosa more susceptible to the carcinogens in tobacco
- B.
Alcohol activates carcinogens present in tobacco by a cytochrome p450-2E1–dependent toxin metabolism mechanism
- C.
Release of free radicals in the mucosa from local and hepatic alcohol metabolism results in mutagenesis.
Studies have shown that heavy alcohol consumption by itself or in addition to tobacco smoking produces about a 5-fold increased lifetime risk for developing oral SCC. Studies looking into the risk associations between alcohol-containing mouth rinses and the development of oral cancer have not revealed any association. This finding was confirmed by an advisory panel convened by the US Food and Drug Administration (FDA) in 1996.
Therefore, although alcohol consumption by itself is not a proven oral SCC causative agent, it is recognized as a potent contributory/independent risk factor when consumed in large quantities and in combination with tobacco smoking.
Human Papilloma Virus
Human papilloma viruses (HPVs) are epitheliotropic DNA viruses with more than 130 identified genotypes. Several strains of HPV (HPV 4,6,11,13,31) are known to cause common viral warts (squamous papilloma, verruca vulgaris) of the skin and mucosal surfaces in children, youths, and adults. The strains HPV 16 and 18 have been closely associated with and implicated in cervical cancers; they are often referred to as the high-risk strains of HPV (HR HPVs). The oncogenic potential of HR HPVs has been attributed to their ability to insert their early genes (E6, E7) into the genome of the host cell, thereby abrogating the critical function of tumor-suppressor and cell-cycle regulatory genes pRb and p53. This situation leads to deregulation of the cell cycle, and impaired apoptotic mechanisms and DNA repair, all of which contribute to malignant progression.
Several recent investigations have reported on the association of HR HPVs and a subset of oropharyngeal SCCs. These studies uncovered a wide range of viral prevalence (0%–100%) in the tumors reviewed. This finding may in large part be attributable to the natural presence of both HR HPVs and other HPV strains in normal mucosa. Nonetheless, in the literature much confusion concerning the role of HPV in oral carcinogenesis has been generated by pooling SCCs from the oral cavity proper and the pharynx and larynx into a single large grouping of head and neck cancer. This tendency to pool disparate site statistics into a single category of disease has led to misunderstanding about the role of HPV relative to carcinogenesis of the oral cavity, because it seems that there are recognized sites in the oropharynx that are more susceptible than the oral cavity mucosa to the oncogenic influence of HPVs. Specific areas in the pharynx and larynx, including the tonsils (ie, squamous-columnar junctions of the crypts and glottides), are believed to harbor greatest susceptibility to HPV because of ready exposure of their basal epithelial cells. (These microenvironments are anatomically similar to the HPV-susceptible transformation zone in the cervix.)
HR HPVs are associated with a subset of SCCs of the pharynx, larynx, and tonsillar tissues in patients who are nonsmokers and nondrinkers. The subset of HR HPVs-associated oropharyngeal SCCs have been shown to be more responsive to radiochemotherapy, and have more favorable treatment outcomes compared with tobacco/alcohol associated SCCs. However, a similar association has not been established between HPV16/18 and oral cavity SCC. Although the presence of E6/E7 HPV transcripts can be noted in some oral SCCs, these early proteins have also been found in perfectly normal oral mucosal epithelial cells. That finding raises the question as to whether HR HPV transcripts present in oral epithelial cells indicate HPV as an agent of carcinogenesis or as an innocent bystander. The tendency to group some HPV-positive oropharyngeal cancers with oral cavity cancers likely accounts for the purported but apparently mistaken notion that oral cancers in general are associated with HPV.
HPV is a common virus associated with many benign oral lesions (verruca vulgaris, squamous papilloma, Heck disease). HPV infection is associated with some oropharyngeal SCCs, but HR HPVs (HPV 16/18) do not seem to play an identified role in oral SCC, and the alleged connection has not been confirmed. Tobacco and alcohol remain the most well-established oral SCC risk factors.
Genetic Susceptibility
The relationship between oral SCC and exposure to tobacco and alcohol is well recognized. It is also well known that only a fraction of smokers and drinkers develop oral SCC. The incidence of oral cancer in individuals less than age 40 years who have no known risk factors has been increasing in recent years and may be attributable to underlying genetic susceptibility. As discussed earlier, individuals with underlying defects of key molecules/genes responsible for maintaining local homeostasis are predisposed to developing oral SCC when exposed to initiating/promoting agents like tobacco and alcohol. Emerging studies show inherent differences between individuals in their DNA repair mechanisms, cell cycle control/apoptosis mechanisms, and/or immune surveillance mechanisms, among others. If future population-based studies successfully identify underlying genetic defects that predispose individuals to developing oral SCC, the information gleaned could substantially improve primary/secondary prevention and early detection.
Radiation
Exposure to ionizing radiation is a well-recognized risk factor for nonmelanoma skin cancers (basal cell carcinomas and SCCs). Ultraviolet (UV) radiation is known to cause DNA damage and mutations in critical DNA repair mechanisms (p53, MDM2) and is a known risk factor for actinic cheilitis and SCC of the lip. However, there is no association between exposure to ionizing radiation and the development of intraoral SCC. Therefore, as discussed earlier, it is important to exclude SCCs of the lip in our discussion of intraoral SCCs. Therapeutic irradiation of the head and neck does not seem to induce second primary SCCs. In years past it was believed that therapeutic irradiation of oral verrucous carcinomas was associated with a high transformation rate to conventional and anaplastic oral SCC. However, more recent analysis reveals this claim not to be true. Therapeutic irradiation of verrucous carcinoma has been shown to yield results comparable with those obtained with conventional SCC.
Clinical considerations: oral cancers and precancerous lesions
Familiarity with the profile of a patient who has oral cancer, including the risk factors as described earlier, and the composite classic clinical features of an oral cancer and its precursor lesions, enhances the clinician’s ability to diagnose and intercept evolving carcinomas.
SCC: Clinical Features
Clinical presentation
Who is the classic patient with oral cancer? Because the process of malignant transformation is notoriously protracted, oral SCC is a disease of individuals 45 years of age or older; it is less common in younger persons (see Fig. 1 ). There is usually a past or active history of tobacco use. Risk for developing a second upper aerodigestive tract malignancy in patients who have oral cancer who continue to smoke is also significantly magnified. The implications of smoking go even further: long considered a male-dominated disease, in recent decades the male/female ratio for oral cancer has been narrowing toward parity. This situation is likely attributable in part to the more socially acceptable use of tobacco among women that began during the period just before World War II and persisted, thus effecting the shifting gender predilection for oral cancer. Tobacco exposure combined with alcohol ingestion seems to heighten the risk for disease. Although moderate intake of alcohol alone was believed to function not as a direct carcinogen but instead as a promoter or potentiating agent for cancer development, recent epidemiologic investigation suggests that chronic ingestion of alcohol at high levels can be an independent risk factor for oral cancer. In unusual cases of oral SCC in individuals less than 40 years of age, epidemiologic studies have not disclosed a significant association between chronic alcohol or tobacco use. Individuals with a prior history of oral cancer or a documented history of one or more precancerous oral lesions are at significant risk for recurrent disease or second primary carcinomas in the oral cavity and upper aerodigestive tract.
Location
Where are oral cancers found? Most SCCs do not occur randomly within the oral cavity. Knowing their most likely locations is vital, not only for case finding (ie, establishing a diagnosis when abnormal signs and symptoms are obvious) but also with regard to screening for oral cancer (ie, pointedly examining for suspicious findings in otherwise asymptomatic individuals or population groups at risk for the disease). Earlier US studies of large numbers of small, localized primary oral SCCs revealed that most of the tumors appeared to favor certain intraoral locations more than others. These so-called oral cancer-prone sites include the lateral and ventral aspects of the tongue, the floor of mouth, and the soft palate-uvula-tonsillar pillar complexes. All of the latter locations are surfaced by unkeratinized mucosa. Presumably, in these particular areas carcinogens dissolved in saliva encounter a thin mucosal barrier that permits their prolonged contact with and ready access to the surface epithelial squamous cells.
Appearance
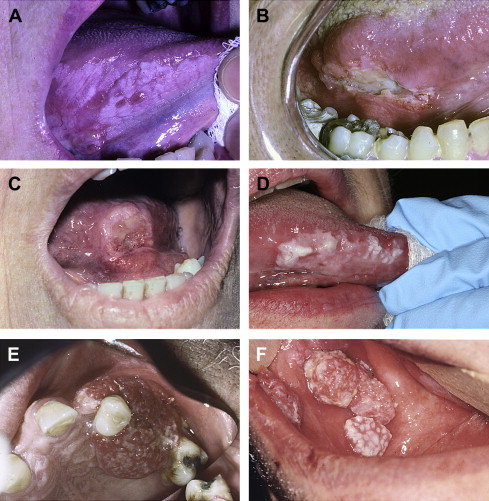
Classic (conventional) oral SCC in most cases presents itself on one of the aforementioned cancer-predilection sites as an ulcerated white, or red, or red and white mass, usually with nonhomogeneous (eg, corrugated, verrucous, pebbly, or nodular) surface topography ( Fig. 2 A–E). There may or may not be pain or paresthesia. On palpation the mass feels indurated; its extent may be difficult to define because of both the endophytic and circumferential growth patterns of the invasive process. Depending on the location, tumor invasion can and frequently does eventuate in loss of mobility or tethering fixation of neighboring anatomic structures in the path of the tumor. This situation can result in functional compromise relative to speech, mastication, and deglutition. Invasion into adjacent or underlying maxillary or mandibular bone can result in unconfined lytic destruction of the alveolar bone and periodontium attended by dramatic tooth mobility. Tumor infiltration of the inferior alveolar nerve is associated with pain and paresthesia ( Fig. 3 ).