Diseases of bone and the maxillary sinus
Overview
This chapter covers the basic anatomy and diseases of the bones of the face and the maxillary sinus. Most diseases of the jaw are odontogenic in origin but the jaws can also be affected by systemic disease and by local non-odontogenic conditions. The clinical and radiological features, pathology and management of non-inflammatory/infective lesions are described. Chapter 4 deals with inflammations and infections of bone.
7.1 Diseases of bone
Normal jaw skeleton
The mandible is formed of a cortex and rather coarse trabecular medulla. A depression into the cortex may form around the submandibular salivary gland during development. It can give rise to a radiolucent area at the angle of the mandible, referred to as Stafne’s cavity (Fig. 7.1). It is important to be aware of this normal structure, which appears below the inferior alveolar nerve canal on radiographs, to avoid confusion with bone cysts. Another important normal structure is the torus mandibularis. Tori are smooth bone prominences found on the lingual aspect of the mandible, below the canine/premolar teeth (Fig. 7.2). They are often bilateral and may consist of single, double or triple prominences. The maxilla is often extensively pneumatised to form the maxillary sinus, described later in this chapter. The hard palate forms by elevation and fusion of embryonic shelves. A bony prominence may form in the midline, which is referred to as torus palatinus. Both the torus palatinus and pterygoid hamulus can be discovered by anxious patients and reassurance may be required.
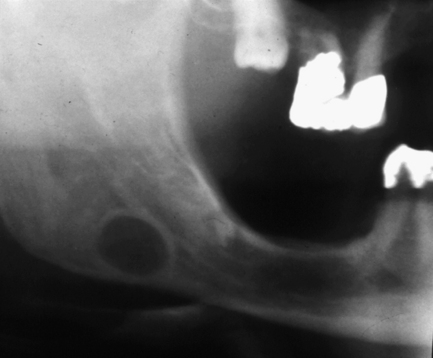
Fig. 7.1 Stafne bone cavity.
This radiograph shows the typical appearance of a rounded well-defined radiolucency with corticated margins, below the inferior dental canal.
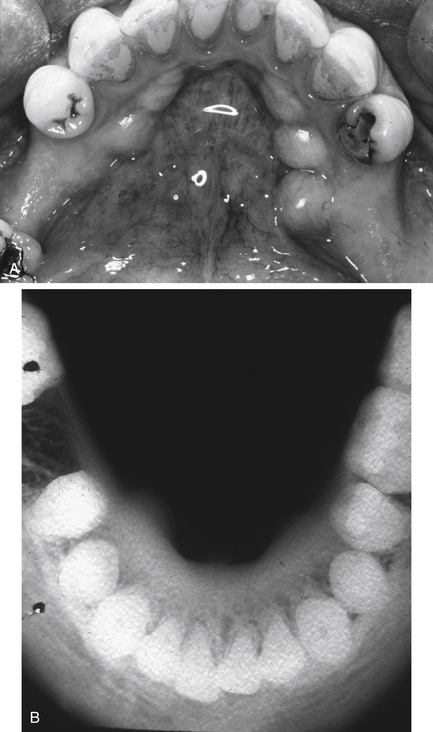
Fig. 7.2 Torus mandibularis.
(A) Clinical appearance. (B) True occlusal radiograph of the mandible showing bilateral protruberances of the lingual cortical bone.
At a histological level, bone is composed of mineralised collagenous matrix containing osteocytes. It is organised into an outer cortex and an inner cancellous (trabecular) structure, which is adaptive to stresses (Fig. 7.3). Endosteal surfaces are lined by bone lining cells; remodelling is achieved by the coordinated activity of osteoclasts (bone-resorbing cells) and osteoblasts (bone-forming cells) in bone metabolic units. Bone is surrounded by periosteum, which is continuous with oral mucosa in certain places in the jaws. The vascular supply to bone is via periosteal vessels and marrow spaces. Fatty and haemopoetic marrow may be present in the jaws.
Fibrous dysplasia
Clinical features
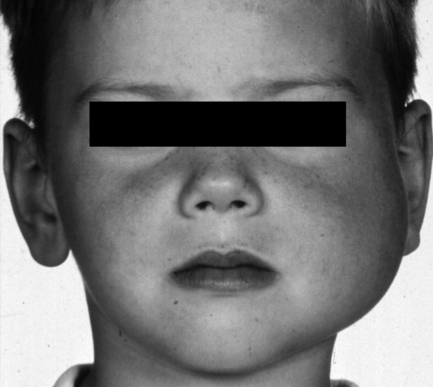
An affected bone or area within a bone undergoes painless expansion. Other symptoms are few, but when the skull base is involved, neurological signs may occur, presumably owing to pressure on foramina. In the jaws, teeth are often affected, with effects upon eruption and developing malocclusion. The maxilla is affected twice as commonly as the mandible. The disease is most commonly unilateral but may involve multiple craniofacial bones and typically produces a visible facial asymmetry (Fig. 7.4). Fibrous dysplasia develops during childhood, usually before 10 years of age, with no sex predilection (except Albright’s syndrome). The disease becomes quiescent in early adult life, but the deformity persists.
The polyostotic form of fibrous dysplasia shares these general characteristics but has additional signs. There are two types: Jaffe’s type and Albright’s syndrome. In the first, multiple bones are affected and there are patches of skin pigmentation (café-au-lait spots). In Albright’s syndrome, which is almost always a disease found in females, there are usually various endocrine abnormalities, such as precocious puberty, hyperthyroidism and hyperparathyroidism.
Pathology
The histopathological appearance is dependent on the stage of disease development. Initially, normal bone is replaced by cellular fibrous tissue within which, as the disease progresses, irregular islands and fine trabeculae of metaplastic woven bone develop. As the lesion matures, so too does the connective tissue, becoming more collagenous, while the bone is remodelled to a lamellar pattern. The lesional tissue merges with the adjacent normal tissue.
Radiology
Initially an affected area appears radiolucent, reflecting the fibrous tissue content. As bone forms, the lesion becomes more radio-opaque. The alteration in trabecular pattern is particularly notable: the trabeculae are very small and fine, resulting in a picture that has been described as like ‘ground glass’, although coarser forms are often described as resembling a ‘fingerprint’ or ‘orange peel’ (Fig. 7.5). Where teeth are present, another commonly noted sign is loss of lamina dura. With age, there is a tendency for lesions to increase their radio-opacity. While lesions classically merge into surrounding normal bone, mandibular lesions sometimes have better defined margins.
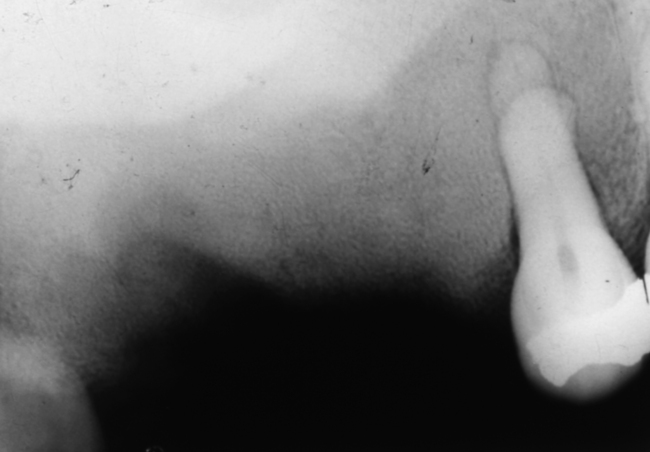
Paget’s disease of bone
Clinical features
Paget’s disease affects individuals in middle and old age. Males are more commonly afflicted than females. The clinical symptoms reflect the enlargement and weakening of bone that results from Paget’s disease. Slowly growing swelling of bones may lead to shape changes and enlargement of the skull and jaws (Fig. 7.6). Deformity of bones, typically of those bearing weight, may lead to bowing of legs and spinal curvature. Bone pain may occur and, if the skull base is affected, various neurological effects may develop.
Pathology
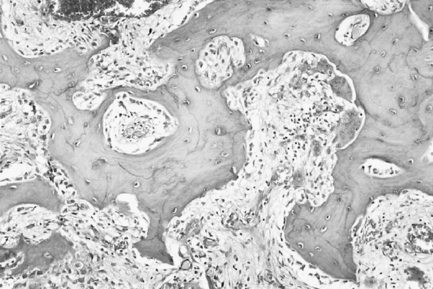
Paget’s disease can be roughly divided into three overlapping phases. During the first of these, osteoclastic activity predominates, normal bone is resorbed and is replaced by well-vascularised cellular fibrous tissue. The surface of the bone is rimmed by giant osteoclasts resting in Howship’s lacunae. As the disease progresses, this osteolysis is accompanied by osteogenesis as new bone forms within the cellular fibrous tissue in the second phase of the disease. This combination of bone resorption and deposition gives rise to the classic mosaic appearance of bone in Paget’s disease (Fig. 7.7). The basophilic reversal lines that outline ‘the pieces of the mosaic’ mark switches in activity from bone resorption to bone deposition. Ultimately, in the third phase, osteoblastic activity predominates and the trabeculae of bone fuse together to give rise to masses of dense, sclerotic bone that is relatively avascular. Cementum is affected by Paget’s disease in a similar manner to bone, resulting in hypercementosis and, when bone and cementum fuse, ankylosis.
Radiology
1. Radiolucent (osteolytic): bone resorption results in radiolucency and cortical thinning; the lamina dura of teeth may disappear.
2. Mixed: the bony trabecular pattern is altered and often appears like ‘ground glass’ or may show a striking appearance of lines with few connections (Fig. 7.8); a few radio-opaque patches may appear in the bone.
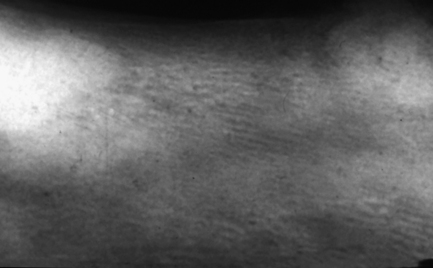
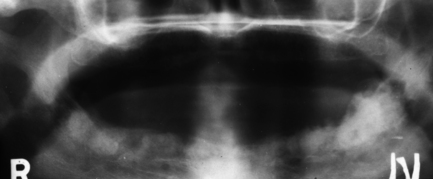
Fig. 7.8 Intraoral radiograph of the mandible of an edentulous patient with Paget’s disease of bone.
There are two main features to note. There is an altered trabecular pattern with an impression of linearity/parallel lines. Mesially and distally there are densely radio-opaque areas (‘cotton wool’ radio-opacities).
3. Radio-opaque (osteoblastic): with time, the radio-opaque patches increase in number, grow and coalesce; tooth roots often have hypercementosis.
The affected bone will always enlarge. In maxillary lesions, the enlargement encroaches on the maxillary sinuses, often obliterating them entirely (Fig. 7.9).
Management
If Paget’s disease is suspected, the serum alkaline phosphatase level should be measured. This is elevated while serum calcium and phosphate levels are normal. Observation may only be appropriate in an elderly patient with no symptoms. However, medical treatment is indicated in those with pain or neurological signs. This consists of calcitonin and bisphosphonates, which inhibit osteoclast activity and slow rather than stop the disease process. Oral surgery should be avoided if possible, and patients given antibiotic cover when it is necessary.
Giant-cell granuloma (central giant-cell granuloma)
Giant-cell granuloma (GCG) is a non-neoplastic lesion of bone.
Pathology
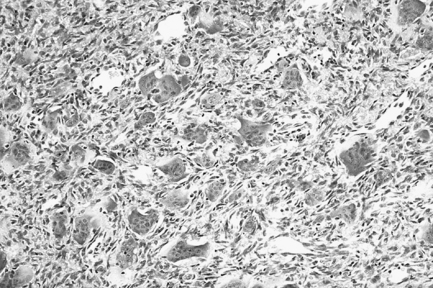
This lesion is identical histologically to the giant-cell epulis (peripheral giant-cell granuloma) and the brown tumours of hyperparathyroidism (see p. 156) and must be distinguished on clinical grounds. GCGs are characterised by the presence of multinucleate osteoclast-like giant cells lying in an extremely vascular stroma. The giant cells vary in size, shape, intensity of staining and the number of nuclei that they contain. The fibroblastic stroma is densely cellular and rich in capillaries, with which the giant cells are often intimately related. Extravasated red blood cells and deposits of haemosiderin may be present. Evidence of dystrophic calcification and metaplastic bone formation may also be seen. In some lesions, fibrous septa delineate foci of giant cells.
Radiology
A round or ovoid radiolucency can be seen with a well-defined, non-corticated margin. Expansion is a common feature, with cortical thinning and sometimes perforation, producing a soft tissue mass. Occasionally there is wispy internal calcification. Displacement of teeth often occurs but resorption is less common (Fig. 7.10).
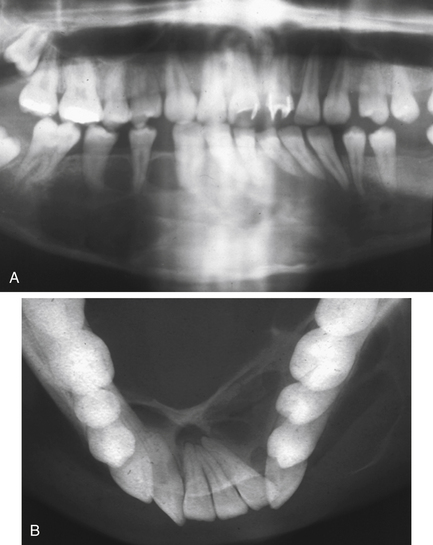
Fig. 7.10 Granuloma.
(A) A panoramic radiograph of a 20-year-old female who presented with a painless swelling of the anterior mandible with displacement of teeth. (B) True occlusal radiograph of the same patient, showing the marked buccal and lingual expansion. The expanded cortices are very thin, suggesting rapid growth.
Management
It is important to distinguish the GCG from hyperparathyroidism. This is normally done by estimating serum calcium, which is raised in hyperparathyroidism. Patients with hyperparathyroidism are referred to a physician for further investigations and treatment. Surgical curettage of a GCG is usually adequate. This treatment may need to be repeated if there is recurrence; sometimes a wider resection may be indicated. Radiotherapy is contraindicated, as with any benign bone lesion.
Osteoporosis
Hyperparathyroidism
Pathology
Cortical bone is more severely affected than cancellous bone. The increase in osteoclastic activity results in thinning of the cortices with loss of lamina dura. Marrow is replaced by fibrovascular tissue; brown tumours of hyperparathyroidism may develop (Fig. 7.12).
Radiology
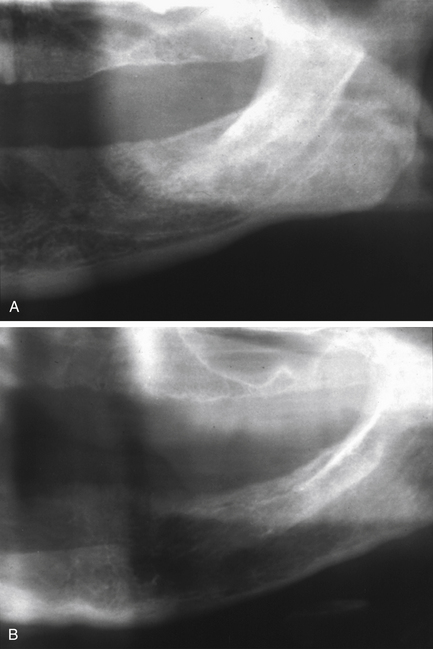
There is increased radiolucency of bone, either generalised or localised. The earliest sign is subperiosteal resorption of the terminal phalanges. In the jaws, lamina dura of teeth is classically lost, along with the cortex of the inferior dental canal. There may be demineralisation of the cortex of the lower border of the mandible. Localised fairly well-defined radiolucencies (brown tumours) may be seen throughout the skeleton but are more common in facial bones than elsewhere (Fig. 7.13).
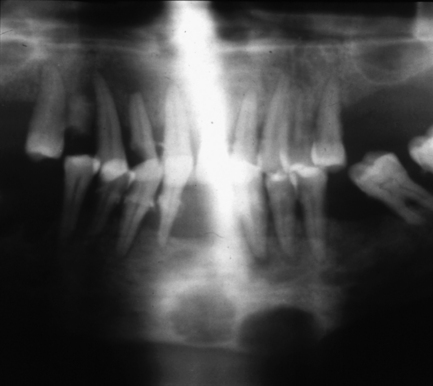
Fig. 7.13 Brown tumours of hyperparathyroidism.
There are two fairly well-defined radiolucencies in the symphysis and parasymphysial region of the mandible; these were brown tumours. Lamina dura is also difficult to identify on the teeth. The radiolucency in 12 region may be inflammatory rather than related to the systemic disease.
Management
If hyperparathyroidism is suspected, assays of serum calcium, phosphate and alkaline phosphatase should be carried out by a physician. Both serum and urinary calcium levels and serum PTH levels are usually raised and serum phosphate levels decreased. Alkaline phosphatase levels are raised in severe disease. The most frequent cause of primary disease is an underlying parathyroid adenoma.
Genetic disorders
Numerous genetic disorders affect the jaws, and a good reference source for evaluation of individual cases is the Online Mendelian Disorders in Man (OMIM) database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim). A number of disorders have effects in the jaw bones:
• Familial adenomatous polyposis (Gardner’s syndrome): multiple osteomas and odontomes, hazy sclerosis and hypodontia may be found in the jaws; numerous polyps develop in the large bowel and there is a very high risk of malignant change (adenocarcinoma of the bowel).
• Osteogenesis imperfecta: multiple bone fractures occur after minor trauma and soft tissues are typically lax, with hernia formation; the sclera may look blue and some patients develop dentinogenesis imperfecta; short stature is typical.
• Osteopetrosis: also known as marble bone disease, the medullary cavity infills with dense bone; the maxillary sinus may fail to pneumatise and bone appears dense and structureless on radiographs; there may be partial failure of tooth eruption.
• Vitamin-D-resistant rickets (hypophosphataemia): description of condition to follow from author
• Cleidocranial dysplasia: description of condition to follow from author
• Cemento-osseous dysplasia: description of condition to follow from author
• McCune–Albright syndrome: description of condition to follow from author
Bone tumours
A simple classification of bone swellings is given in Box 7.1. Osteomas occur most frequently in the paranasal sinuses and are dealt with in the section on maxillary sinus. Primary malignant bone tumours are rare and include osteosarcoma, chondrosarcoma and multiple myeloma. Direct invasion of bone by squamous-cell carcinoma arising in the oral mucosa is common in advanced oral cancers. Metastatic deposition of carcinoma from colon, lung, breast, kidney and other primary sites is more likely to be the cause of a destructive malignant lesion in bone than primary sarcoma.
7.2 Diseases of the maxillary sinus
• patients with maxillary sinusitis or other pathoses may present to the dentist believing they have toothache
• patients with dental pathology in the maxilla may develop secondary signs and symptoms in the sinus
• dentists may intrude into the maxillary sinus during surgery or other dental procedures.
Anatomy
The normal sinus in adults is pyramidal in shape. At birth, it is very small, growing laterally from its point of origin above the inferior turbinate bone until, by about the ninth year, it extends to the zygoma. Lateral growth ceases by 15 years of age. Radiologically, the sinus appears as a triangular-shaped air containing cavity on cone beam computed tomography (CBCT) and CT images (Fig. 7.14). On intraoral radiographs, the antrum is demarca/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


