Biocompatibility
 Critical Question
Critical Question1. The chemical nature of its components
2. The physical nature of the components
3. The types and locations of patient tissues that will be exposed to the device
4. The duration of the exposure
5. The surface characteristics of the material
6. The amount and nature of substances eluted from the material
 Critical Question
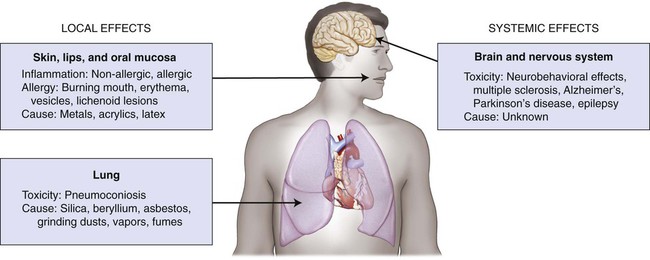
Critical QuestionA critical adverse effect is the first event that is observed at the lowest exposure level. The location of this effect is called the critical tissue, or critical organ, and the concentration of a substance that produces this effect is the critical concentration. A metal such as mercury can be exposed to tissues as a solid binary phase (Ag2Hg3), as dissolved ions in saliva, and as atoms in the vapor form. For this metal, one must be concerned about exposure levels, absorbed dose, body burden, and critical target-tissue concentrations. Shown schematically in Figure 7-1 are the critical tissue and organ sites that can be affected by exposure to dental restorative materials and auxiliary materials used to make impressions and models. Material components can be released during melting and casting of metals, fabrication of prostheses, grinding and polishing procedures, adhesive bonding, or cementing to prepared teeth. Exposure by deposition and possibly by local uptake occurs when any one of the four types of epithelial surfaces—oral tissue, gastrointestinal tract tissue, respiratory tract tissue, and skin—makes contact with one of these metal forms.
Traditionally, we have accepted the view that toxicity is dose dependent and allergy is dose independent. However, there is considerable uncertainty about which types of exposures lead to the sensitization of individuals to substances or ions released from dental restorative materials and auxiliary dental materials. Recent research suggests that dose effects for food allergies may be meaningful in describing the severity of allergic reactions. One of the common descriptors used is the dose corresponding to a 10% increase in an adverse effect relative to the control response. This is designated by ED10. An analysis of a database for 450 subjects with peanut allergy indicated that the ED10 was 12.3 mg of whole peanut (average mass of 1000 mg per whole peanut) compared with a 5% increase (ED05), corresponding to an ingested mass of 5.2 mg (Taylor et al., 2012). Thus, it is clear that sufficient data exist to establish a threshold level for a peanut allergy. However, there are no similar types of data for comparative evaluations of dose effects on the severity of allergic reactions to restorative materials. Instead, we must rely on the signs and symptoms expressed by our patients and the visual signs observed during clinical exams. In certain cases, allergy tests may have to be performed by an allergist or dermatologist (if appropriate) to assist in the diagnostic process. Specific dental test substances have been established for this purpose, and the treating dentist may have to make additional suggestions to the allergist for tests of other dental substances that are of potential significance in each case (Gawkrodger, 2005; Khamaysi et al., 2006).
Historical Perspective
 Critical Question
Critical QuestionTests for the safety of restorative dental materials must ensure that a candidate material is nontoxic and unlikely to cause adverse immunological effects. Evaluations of toxicity are designed to identify adverse health events caused by physical agents, chemical agents, or both. Paracelsus (1493−1541) correctly proposed that only the dose of a substance differentiates a toxic agent from a remedy (Siddiqui et al., 2003). No pharmacological agent is free of potential toxic effects, and no drug is free from the possibility of causing an adverse event in certain individuals. No test can produce results that can guarantee that a substance will not cause adverse effects in all individuals who are treated with the substance. The allowable percentage of adverse effects in a population is based on the risks to the health and life expectancy of the individuals who will be exposed to the product under the indicated conditions and the corresponding exposure doses for its components.
Requirements of the U.S. Food and Drug Administration
1. Class I: Low risk—General Controls [generally exempt from 510(k)]
2. Class II: Moderate risk—General Controls & Special Controls [510(k) generally required]
3. Class III: High risk—General Controls & Premarket Approval (PMA) Application (PMA required; must demonstrate safety and effectiveness without relying on a predicate device)
A brief listing of FDA device classifications and applicant requirements are summarized below.
510(k) Classification
The most common pathway to market for medical devices
A review to determine whether a new device demonstrates substantial equivalence (SE).
SE to a predicate device (pre-amendment or post-amendment) for which PMA is not required
FDA’s determination of SE serves as the classification process for new devices
Most of the submissions to the Dental Branch are 510(k) types of submissions
Substantial Equivalence / Substantially Equivalent
A new device is deemed SE to a predicate device if:
1. It has the same intended use;
2. It has the same technological characteristics (includes chemistry, materials, design specifications, mechanical, or biological properties, etc.); or
3. It has different technological characteristics, but it does not raise new types of safety and effectiveness questions and is at least as safe and effective as the predicate device.
Adverse Effects from Exposure to Dental Materials
 Critical Questions
Critical QuestionsLocal and Systemic Effects of Materials
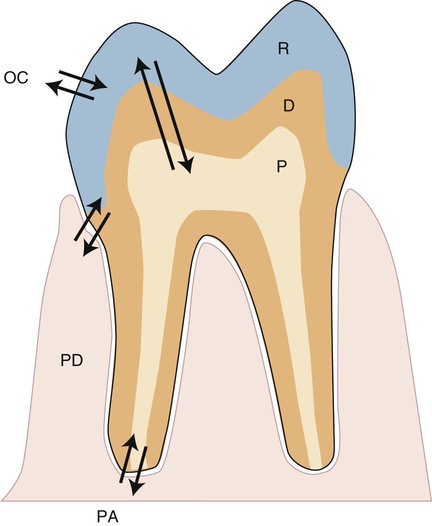
Any biomaterial that is placed adjacent to a natural tissue in the body can induce local or systemic biological effects. These effects are controlled by the substances that are released from the material and the biological responses to those substances. The nature, severity, and location of these effects are determined by the distribution of released substances. For dental materials, local effects might occur in the pulp tissue, in the periodontium, at the root apex, or in nearby oral tissues such as the buccal mucosa or tongue (Figure 7-2). The arrows in this figure indicate the pathways that foreign substances from a restorative material, if present, take into the oral environment, the tissue space next to the periodontium (PD), the pulp chamber (P), or the periapical region (PA). The periodontal ligament is also an important tissue, since it is located in proximity to the pocket or attachment area, which is often a site for accumulation of biofilms and ions, atoms, or molecules of substances released from the cervical region of dental restorations that can extend into this area. Such accumulations can be metabolized, which could then change their biological properties. These local effects are a function of (1) the ability of substances to be distributed to these sites, (2) their concentrations, and (3) exposure times, which may range from seconds to years.
Inflammatory and Allergic Reactions
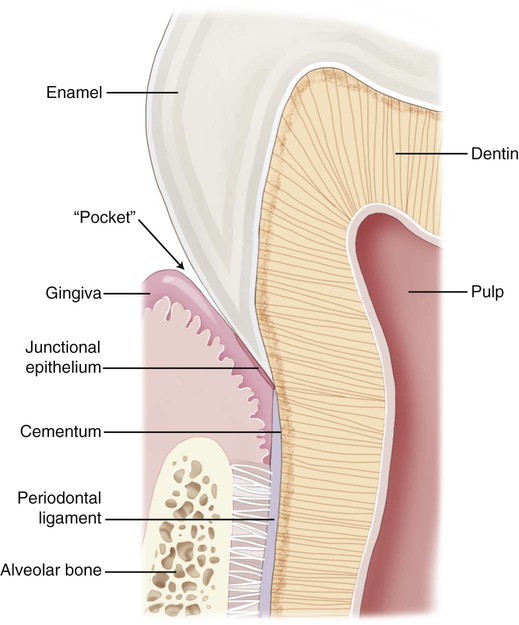
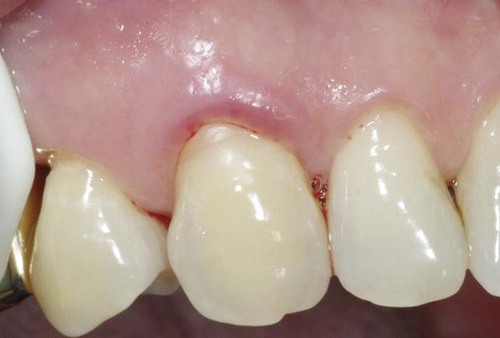
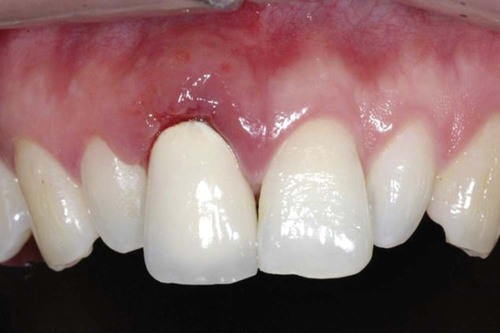
Different types of biological responses to substances can occur in humans. These include inflammatory, allergic, toxic, and mutagenic reactions. However, not all of these have been documented for dental material exposures. The inflammatory response involves the activation of the host’s immune system to ward off some challenge or threat. Inflammation may result from trauma (excessive force, laceration, and abrasion), allergy, or toxicity. Histologically, the inflammatory response is characterized by edema of the tissue caused initially by an infiltration of inflammatory cells such as neutrophils and, later in the chronic stage, to the action of monocytes and lymphocytic cells. The relationship of dental materials to inflammatory reactions is important because of chronic inflammatory responses such as pulp inflammation and periodontal disease. As indicated previously, teeth with cervical restoration margins can release ions or other substances into the gingival sulcus, and adverse reactions can affect the periodontal attachment and the periodontal ligament (Figure 7-3, with no restoration).
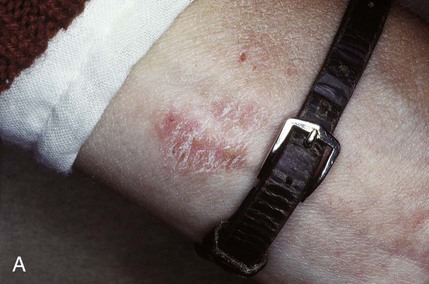
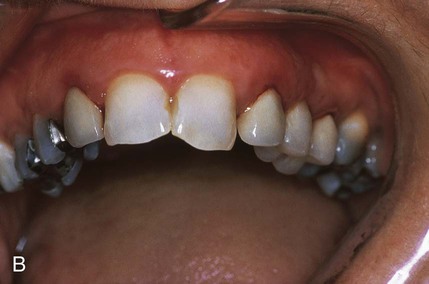
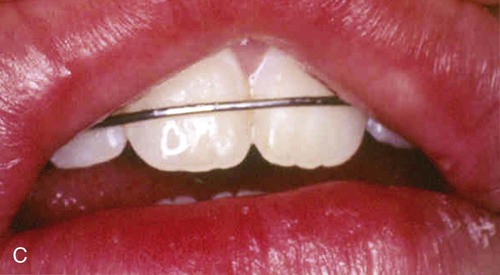
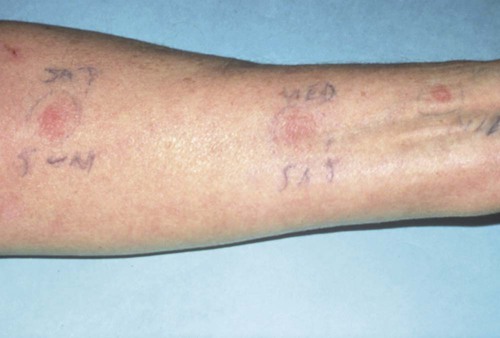
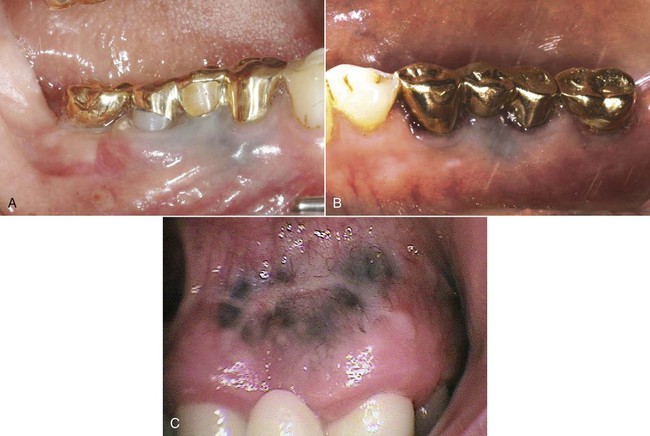
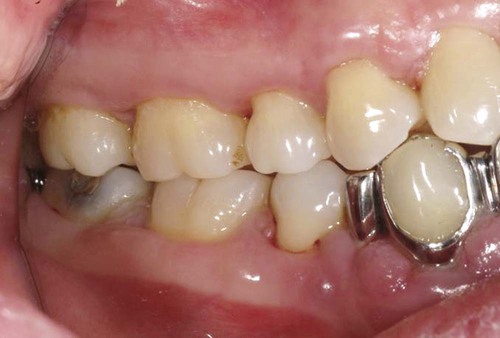
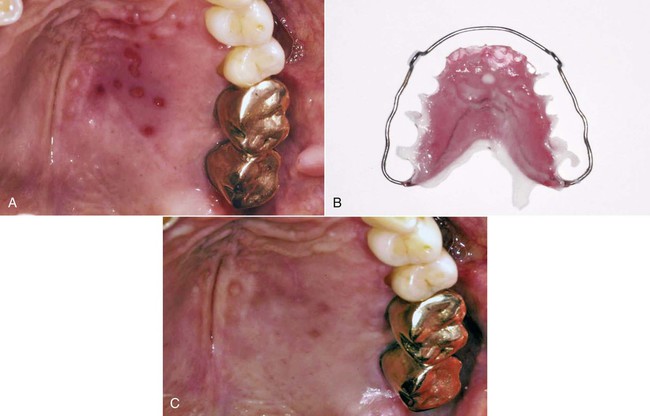
An allergic reaction induces an inflammatory response that cannot easily be distinguished from reactions caused by a nonallergic inflammatory process or by low-grade toxicity. However, by observing the signs of the effect or the absence of the signs in other locations and by the process of elimination, some reasonably logical inferences may be drawn. In some cases, observation for 2 weeks or more, when possible, can lead to a resolution of the response because the effect was caused either by trauma, another noninflammatory process, or a self-limiting allergic condition. Examples of inflammatory reactions that may be caused by allergens leached as ions from metals or other substances released by dental materials are shown in Figures 7-4 through 7-10. Figure 7-5 illustrates three potential sites for allergic reactions to nickel-containing metals: (1) a watchband buckle; (2) bilateral, fixed metal-ceramic prostheses with copings and framework made from nickel-alloy (crown on left and three-unit FDP on right); and (3) a severe reaction of lips to nickel-containing wire. Figure 7-5, D, illustrates positive responses to patch-test substances on a patient’s back. In addition, bilateral lichenoid mucositis lesions which will be discussed later, are shown in Figure 7-11 on the buccal mucosa adjacent to gold alloy crowns.

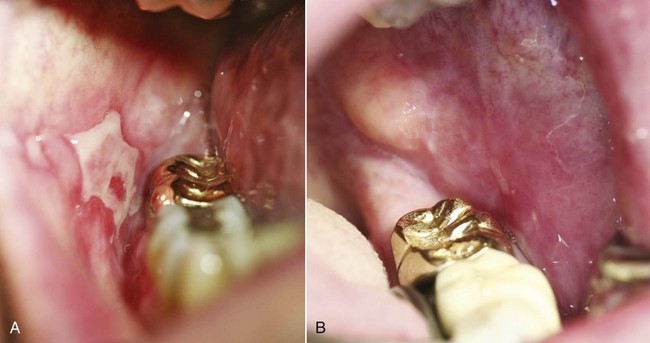
Some materials, such as latex, can cause allergy directly by activating antibodies to the material. These are classified as Type I, II, or III reactions, according to the Gell and Coombs classification of immune responses (Gell and Coombs, 1963; Rajan, 2003). These reactions occur quickly and are modulated by antibody-producing eosinophils, mast cells, or B lymphocytes. In comparison, metal ions must first interact with a host molecule to produce a delayed Type IV hypersensitivity reaction, which is modulated by monocytes and T cells. This type is often associated with contact dermatitis.
A Type II response is a cytotoxic hypersensitivity reaction, Type III is an immune complex hypersensitivity reaction, Type IV is a delayed or cell-mediated hypersensitivity, and Type V is a stimulating-antibody reaction, which is rare and sometimes classified as a subcategory of Type II (Rajan, 2002).
 Critical Question
Critical QuestionPrecursors to Adverse Effects of Dental Materials
 Critical Question
Critical QuestionProducts that pass the primary tests, such as the toxicity test, then progress to secondary and usage tests (Figure 7-12). Today, the choice of the test method is based on risk assessment, which is also divided into separate stages of analysis. Details on such testing are provided in relevant ISO standards such as ISO 14971. This risk assessment is the basis for deciding whether new tests are necessary or not, or whether data from the scientific literature are sufficient. If they are necessary, cell culture tests are regarded as the first choice combined with animal tests for sensitization. Subsequently, risk assessment continues and decisions are made on whether or not further tests (e.g., animal tests or human clinical tests) are needed. Clinical tests in some countries are mandatory for materials that are considered to be of high risk. The final decision for market clearance is then made by an interchange between manufacturers and a third party such as a governmental agency or a private organization to which this authority has been granted by a government agency. Our profession seems to be overly complacent in its acceptance of new materials without demanding proof of their safety and efficacy. When a new product is introduced on the dental market, the advertisements tend to promote the clinical performance but rarely summarize the biocompatibility tests and results of these tests compared with results from control materials.
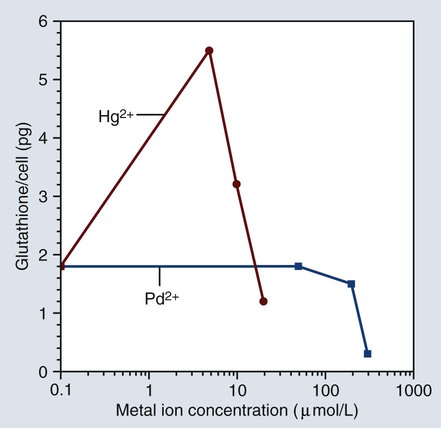
Since toxicity is dose dependent, it is obvious that materials that release too much of a substance can cause overt toxicity. The terms and definitions given previously indicate that there are different dose thresholds for various levels and probabilities of risk. Figure 7-13 shows a plot of cellular glutathione from monocytes that were exposed to mercury or palladium ions in a cell culture. Mercury ions are shown to increase the glutathione content of the monocytes in the cell culture, while palladium ions cause the glutathione content to decrease. Since glutathione is essential for maintaining the redox balance in the cells, exposure to these metallic ions can change the cellular function of the monocytes.
Immunotoxicity
Our understanding of the differences between toxicity, inflammation, allergy, and mutagenicity has become clearer as we learn more about the interactions between biomaterials and cells. The principal concept of immunotoxicity is that substances leached from materials can alter immune system cells, resulting in enormous biological consequences because of the amplifying nature of immune cells. These cellular alterations can occur initially because of the toxic effect of a leached substance. Monocytes control chronic inflammatory and immune responses, and they also secrete many substances that alter the actions of other cells. Thus, if substances leached from a biomaterial change the monocyte’s ability to secrete these substances, the biological response can be greatly influenced and this may greatly impair cellular defense mechanisms against bacteria (Schmalz et al., 2011).
As stated earlier, cell function can either increase or decrease as a result of immunotoxic effects. As shown in Figure 7-13, mercury ions are known to increase the glutathione content of human monocytes in cell culture, whereas palladium ions decrease the cells’ glutathione content. Glutathione is important in maintaining oxidative stress in cells, and any change in its concentration can alter cell function. Higher concentrations of mercury can also decrease glutathione as the ion concentration becomes more toxic.
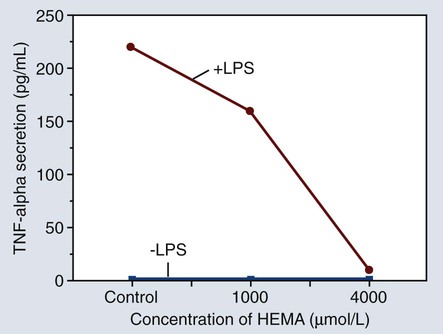
Figure 7-14 shows a graph of the amount of tumor necrosis factor alpha (TNF-α) secreted by monocytes after exposure to hydroxyethyl methacrylate (HEMA) in different concentrations. The +LPS line represents the effect of cell stimulation by lipopolysaccharide while the –LPS line indicates that the monocytes were not stimulated by lipopolysaccharide. The significance of this effect is that relatively small amounts of HEMA released from bonding adhesives or resin-based composites can alter the normal functions of monocytes, thereby contributing to the potential immunotoxicity of some resin-based products.
Pulpal and Periodontal Effects
Because the cervical margins of many dental restorations are near the periodontal attachment area (see Figure 7-3), the biocompatibility of these materials may influence the body’s ability to defend against bacteria that cause periodontal disease. Further, the periodontal pocket, or gingival sulcus, may accumulate significant concentrations of leached substances that do not accumulate to these levels in other areas. In addition to the accumulation of leached or dissolved substances in subgingival areas, substances that are leached from root canal filling materials may accumulate next to the apical foramen. Substances that accumulate in these areas can lead to inflammatory reactions, allergic reactions, periodontal pathology, and periapical lesions.
 Critical Question
Critical QuestionInterfaces with Dental Materials
As shown in Figure 7-15, incomplete bonding or resin penetration into the collagen mesh of acid-etched dentin can lead to fluid ingress along gaps wider than 1 µm, which is referred to as microleakage. The microscopic gap for the so-called microleakage process may lead to several undesirable events. The bacteria that migrate to the pulp may initiate an infection of pulp tissue. The gap also promotes material breakdown along the unsupported margin. This breakdown increases the gap width, which allows larger particles and molecules to progress toward the pulp chamber. It also causes marginal staining and compromised esthetics that may lead to premature replacement. One in vitro study revealed less leakage for amalgam restorations compared with resin-bonded composites (Őzer et al., 2002).
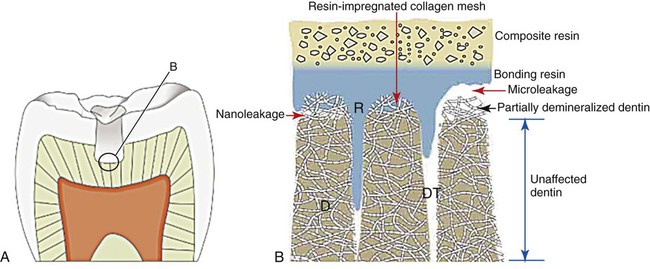
If the resin penetrates the collagen network of dentin but does not penetrate it completely, then a much smaller gap (less than 0.1 µm in most cases) will exist between the mineralized matrix of dentin and the collagen–resin hybrid layer (see Figure 7-15). This much smaller gap has been claimed to allow nanoleakage, which probably does not allow bacteria or bacterial products to penetrate the marginal gaps of the restoration and the pulp. However, fluid exchange most likely occurs, and this may degrade the resin or the collagen network that is incompletely embedded with the resin, thereby reducing the longevity of the dentin–resin bond.
 Critical Question
Critical QuestionInfluence of Biocompatibility on the Osseointegration of Implants
The success of endosseous dental implants is based on the biocompatibility of the implant surface and the ingrowth of new bone into the surface through the process of osseointegration. Very few implant materials or implant coatings promote osseointegration. The most common implant materials include (1) CP Ti; (2) titanium-aluminum-vanadium alloy; (3) tantalum; and (4) some types of ceramics. Materials that allow osseointegration have very low degradation rates, and they tend to form surface oxides that enhance bony approximation. Some materials such as bioglass ceramics promote a perfect osseointegration of the bone. The general process of biointegration involves the adaptation of bone or other tissue to the implanted material without any intervening space along the tissue-material interface (see Chapter 20).
Mercury and Amalgam
Several studies have shown that amalgams release sufficient vapor to cause absorption of between 1 and 3 µg/day of mercury, depending on the number and size of amalgam restorations present (Langworth et al., 1988; Berglund, 1990; Mackert and Berglund, 1997; Ekstrand et al., 1998). The inhaled mercury gains access to the bloodstream via the alveoli of the lungs. From the blood, mercury is distributed throughout the body, with a preference for fat and nerve tissues. Mercury is also ingested as particles produced by wear, and about 45 µg/day of mercury may reach the gut either as the amalgam form or as dissolved and released Hg2+ ions. The absorption of ionic mercury is also poor (approximately 1% to 7%). Mercury trapped in amalgam particles is also poorly resorbed. Methyl mercury is not produced from amalgams but is generally a product of bacteria or other biological systems acting on metallic mercury. Methyl mercury is the most toxic form of mercury and is also very efficiently absorbed from the gut (90% to 95%). Methyl mercury is absorbed mainly from the diet, particularly from fish (especially shark, swordfish, and tuna), which contribute a significant portion.
Numerous studies have attempted to determine whether mercury exposure from dental restorations or other sources contributes to any documentable health problem. Several studies have estimated the number of amalgam surfaces needed to expose an individual to mercury concentrations with a minimum observable effect (slightly impaired psychomotor performance, detectable tremor, and impaired nerve conduction velocity). Estimates are that several hundred amalgam surfaces would be necessary to achieve these levels. Even if all 32 teeth were restored on all surfaces with amalgam, the total number of surfaces (counting incisal edges) would be only 160. Other studies have measured renal function in patients in whom all of the amalgam was removed at the same time (the worst possible case). Despite markedly elevated blood, plasma, and urine levels of mercury, no renal impairment was noted. Still other studies have attempted to look at blood cell types and cell numbers in dentists, who are presumably exposed to higher levels of mercury because of their daily occupational exposure. No effects of mercury have been noted. Other studies for neurological symptoms in children populations occupationally exposed have shown no effects (Bellinger et al., 2006, 2007; DeRouen et al., 2002, 2006). In summary, there are simply no data to show that mercury released from dental amalgam is harmful to the general population.
Methacrylates and Resin-Based Composites
The best screening substance for methacrylate allergy caused by dental material products is HEMA. This result confirms previous findings (Goon et al., 2006), which revealed that HEMA alone picked up 96.7% of the patients with methacrylate allergy and 100% of the dental personnel with methacrylate allergy.
The frequency of positive responses to the common allergen test substances were reported as follows: gold sodium thiosulfate, 14.0%; nickel sulfate, 13.2%; mercury, 9.9%; palladium chloride, 7.4%; cobalt chloride, 5.0%; and HEMA 5.8% (Goon et al., 2006).
Estrogenicity
In 1996, a research group claimed that dental sealants released estrogenic substances in sufficient quantities to warrant concern (Olea et al., 1996). Since then, the estrogenicity of dental composites has also been questioned, particularly for use in children. Estrogenicity is the ability of a chemical to act as the hormone estrogen does in the body. If these chemicals are not indigenous to the body, the substance is called a xenoestrogen. The occurrence of xenoestrogens in the environment has been a concern for many years. Environmentalists fear that these substances will alter reproductive cycles and developmental processes in wildlife, and there is evidence to support these concerns with regard to humans. The concern about estrogens in dentistry centers around a chemical called bisphenol A (BPA), which is a synthetic starting point for bis-GMA (bisphenol-A-glycidyldimethacrylate) composites in dentistry as well as many other plastics. The fear is that the release of these substances might alter normal cellular development or cell maintenance if BPA has estrogenic effects, even at the impurity levels that may be encountered in practice. It is generally accepted that BPA is released from bis-DMA, which is also used in dental products. However, if bis-DMA is used, the amount released after placement of a restorative filling is too small to be of concern. However, to overcome any concerns, products free of bis-DMA can be used.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


