Bone Diseases and Fibro-osseous Lesions
Fibro-Osseous Lesions
II Reactive (dysplastic) lesions arising in the tooth-bearing area
III Fibro-osseous neoplasms: Cementifying fibroma, ossifying fibroma or cemento-ossifying fibroma
Periapical Cemento-osseous Dysplasia
Focal Cemento-osseous Dysplasia
Florid Cemento-osseous Dysplasia
The term florid cemento-osseous dysplasia (FCOD) was first suggested by Melrose et al in 1976 to describe a condition of exuberant multiquadrant masses of cementum and/or bone in both jaws and in some cases, simple bone cyst-like lesions in affected quadrant. Sometimes familial tendencies have been observed. In past this condition has been designated as sclerosing osteitis, multiple enostoses, diffuse chronic osteomyelitis and gigantiform cementoma. The etiology of florid cemento-osseous dysplasia is unknown. Waldron et al have proposed that reactive or dysplastic changes in the periodontal ligament might be a cause for the disease.
Gigantiform Cementoma
Management and prognosis
The surgical management of gigantiform cementoma is usually difficult because of the extensive involvement of the jaws with these tumors. Surgical inaccessibility, particularly in the posterior regions of the jaws seems to be responsible for recurrence of the tumors. These lesions seem to have a tendency toward recurrence when they are treated with incomplete surgical removal. It is therefore recommended that these lesions should be managed with conservative but complete surgical excision whenever feasible.
Ossifying Fibroma, Cementifying Fibroma and Cemento-ossifying Fibroma
Radiographic features
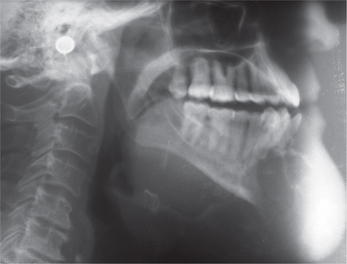
The lesion appears well circumscribed in contrast to fibrous dysplasia, the borders of which are ill-defined. Initial lesions are radiolucent representing an osteolytic image followed by gradual transformation into a mixed lesion and eventually becoming radiopaque. The periodontal ligament space of the involved teeth is clearly seen unlike fibrous dysplasia. Eversole et al have described two basic radiological patterns: a unilocular radiolucency with or without radi-opaque foci, and a multilocular radiolucency. The former presentation is found to be more common. Expansion of the cortical plates is a common finding. Teeth displacement and root resorption may be seen (Figure 1).
Histopathologic features
Ossifying fibroma shows a range of histologic appearances. It shows fibrous and osseous tissues with the former tissue predominating. The fibrous stroma is highly cellular, with spindle-shaped fibroblastic cells arranged in whorls. A fibrous capsule may be seen in some cases. The osseous tissue consists of rounded or lobulated basophilic masses (cementum-like), trabeculae of osteoid, woven or lamellar bone, or combination of the two. Osteoblastic rimming around the trabeculae can be seen. Focal clusters of giant cells (osteoclasts) can be seen to be arranged haphazardly or adjacent to the mineralized material.
Juvenile Ossifying Fibroma (Psammomatoid and Trabecular)
Histopathogic features
Microscopic examination of psammomatoid juvenile ossifying fibroma shows well demarcated but unencapsulated lesion composed of numerous small rounded mineralized collagenous bodies (psammomatoid ossicles) uniformly distributed within a cellular fibroblastic stroma. The cellularity of the fibroblastic stroma varies in different tumors and at different sites within the same lesion. Occasional shrunken cells, representing nuclei of osteocytes, are embedded within the ossicles, and there is usually a collagen outer band to these mineralized bodies. Some of the ossicles may show a basophilic center and eosinophilic fringe. In the periphery of the lesions, some of the ossicles show a transition into small bone trabeculae. The tumor infiltrates and destroys the adjacent bone with focal induction of reactive bone formation. Development of aneurysmal cyst is followed by focal myxoid change in the stroma with hemorrhage and osteoclastic giant cells, with gradual expansion and formation of cysts with thin fibrous walls. The differential diagnosis with other fibro-osseous lesions of the jaw, such as cemento-ossifying fibroma, osteoid osteoma or bone dysplasia, should be made with a mandatory pathological study, and is largely based on the nature of the calcified products of the tumor. The mineralized tissue of central ossifying fibroma is composed of a variable combination of mature and immature bony trabeculae and lobulated basophilic masses of cementum-like material. There may be occasional concentrically laminated particles, called cementicles, but these are not a prominent feature of psammomatoid entity.
Fibrous Dysplasia
Etiology
The molecular mechanism responsible for fibrous dysplasia is a postzygotic activating mutation of the GNAS1 gene that encodes for the Gsα subunit of the heterotrimeric G protein complex. The result is constitutive activation of the adenylyl cyclase enzyme and overproduction of 3′,5′-cyclic adenosine monophosphate. The most common GNAS1 gene mutations are a replacement of arginine by either cysteine or histidine at codon 201 (R201C or R201H), but other mutations have also been identified. The severity of the disease phenotype is thought to depend on when the mutation occurs during embryogenesis. If the mutation occurs during the formation of the inner cell mass, all three germ cell layers will be affected and the phenotype will be McCune–Albright’s syndrome. If it occurs later in development, only one or two germ cell layers will be affected and the phenotype is less severe. Fibrous dysplasia is considered a disease of cells of the mesenchymal stem cell/osteoblastic lineage in which excess cyclic adenosine monophosphate impairs the ability of the stem cell to differentiate into a mature functioning osteoblast.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


