Malignancies of the Jaws
Malignant nonodontogenic tumors of the jaws, both primary and metastatic, are rare in comparisons of soft tissue and mucosal malignancies. Despite their infrequent occurrence, diagnosis of a malignant jaw tumor has serious prognostic implications, often requiring oncologic surgery, radiation, and/or chemotherapy. Generally, these tumors cause signs and symptoms that often are highly suggestive of malignancy (Box 14-1). Tumors discussed in this chapter are those arising from the hard tissues (osteosarcoma and chondrosarcoma) and those nonosseous tumors that frequently involve the mandible and the maxilla (Ewing’s sarcoma, Burkitt’s lymphoma, plasma cell malignancies, and metastatic carcinoma).
Osteosarcoma
Osteosarcomas account for approximately 20% of all sarcomas and, after plasma cell neoplasias, are the most common primary bone tumors. Approximately 5% of osteosarcomas occur in the jaws, with an incidence of less than 1 case in 1.5 million persons per year (Box 14-2). Osteosarcomas can arise de novo or in the setting of several preexisting bone abnormalities such as Paget’s disease, fibrous dysplasia, multiple osteochondromas, bone infarct, chronic osteomyelitis, and osteogenesis imperfecta. Osteosarcoma can also arise in two cancer susceptibility syndromes: hereditary retinoblastoma (RB) and Li-Fraumeni syndrome. Patients with RB inherit a mutation of the retinoblastoma gene on one chromosome and then develop a second mutation of the other retinoblastoma allele. Affected patients develop retinoblastoma of the eye and have an increased risk of sarcoma such as osteosarcoma later in life. Patients with Li-Fraumeni syndrome inherit germline mutations of the p53 gene and have an increased risk of developing a variety of tumors including osteosarcoma. Some osteosarcomas have been preceded by radiation therapy to the affected bone for unrelated or antecedent disease. A vast majority of osteosarcomas involve the long bones, especially in the knee. Osteosarcomas can also be classified by their site of origin as (1) the conventional type, arising within the medullary cavity; (2) juxtacortical tumors, arising from the periosteal surface; and (3) extraskeletal osteosarcomas, arising in soft tissue.
Osteosarcomas are characterized by complex chromosomal aberrations in both number and structure. Gains of 8q23 are present in up to 50% of tumors and correlate with poor prognosis. Increased copy number of the 8q24 region containing the MYC gene has been identified in up to 40% of tumors. Despite the complex karyotype, no specific chromosomal translocation has been identified in osteosarcoma. Alterations in several genes have been consistently identified in osteosarcoma and may contribute to development of the tumor. These include amplifications of MET, FOS, SAS, MDM2, CDK4, and PRIM1. Protein expression of defective/amplified genes results in loss of control of cell proliferation and differentiation (see the discussion on pathogenesis of squamous cell carcinoma in Chapter 2).
Clinical Features
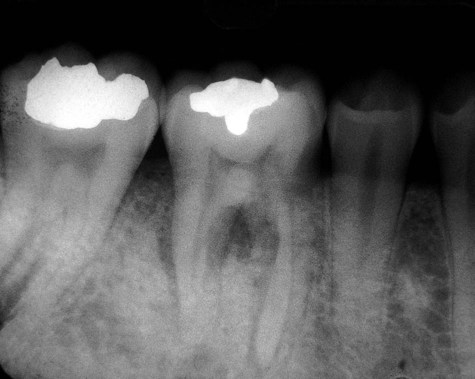
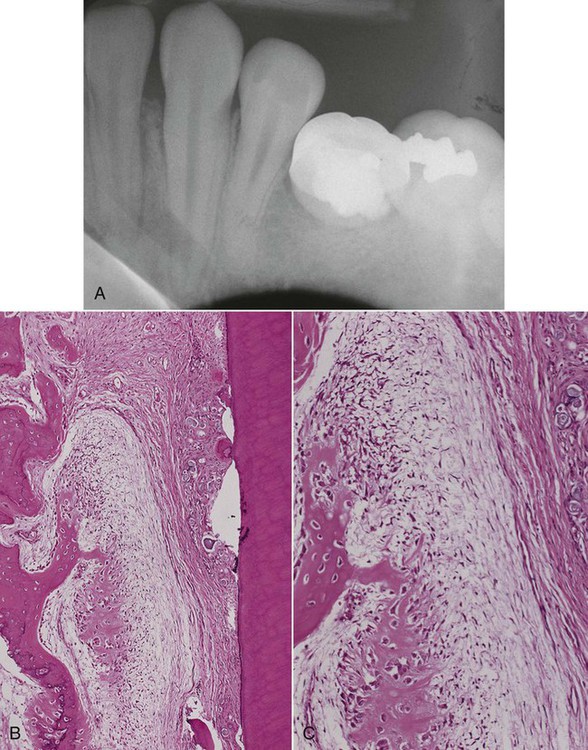
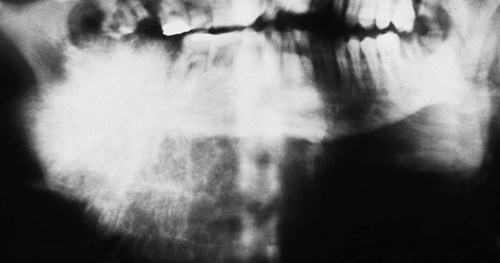
The radiographic appearance of conventional (intramedullary) osteosarcoma is variable, reflecting the degree of calcification of the tumor. There appears to be little relationship between the radiographic pattern and the histologic subtype of osteosarcoma. Early osteosarcomas that involve the alveolar process may be characterized by localized widening of the periodontal ligament space of one or two teeth (Figures 14-1 and 14-2). The widened space results from tumor invasion of the periodontal ligament and resorption of surrounding alveolar bone (Figure 14-3). Advanced tumors can be visualized as “moth-eaten” radiolucencies or as irregular, poorly marginated radiopacities. Most of these neoplasms have mixed radiographic features. A characteristic “sunray” or “sunburst” radiopaque appearance due to periosteal reaction may be seen in jaw lesions but is not diagnostic of osteosarcoma (Figures 14-4 and 14-5).
Histopathology
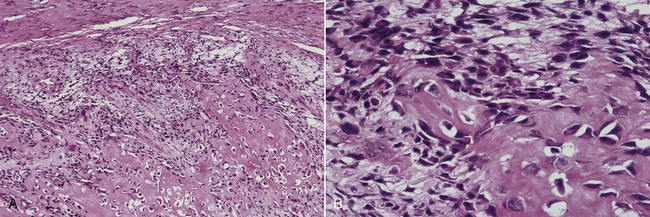
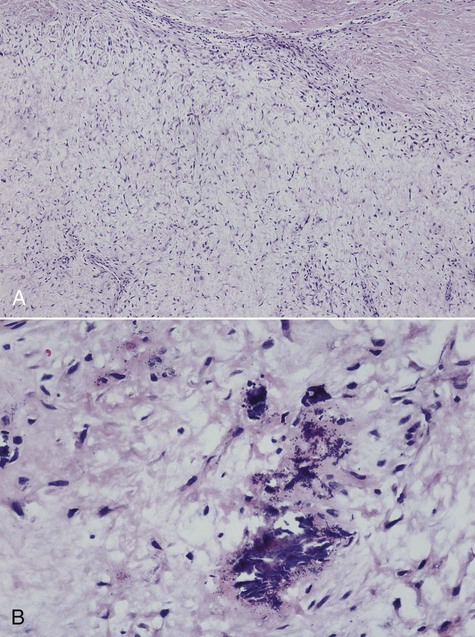
Microscopically, all osteosarcomas have in common a sarcomatous (malignant spindle cell) stroma that directly produces osteoid (Figures 14-6 and 14-7). Histologic subtypes are recognized and have been designated as chondroblastic when formed malignant cartilage predominates (most common) (Figure 14-8), osteoblastic when malignant bone and osteoid predominate, and fibroblastic when spindle cells predominate (Figure 14-9). An additional variant, designated as telangiectatic, contains multiple blood-filled aneurysmal spaces lined by malignant cells but rarely occurs in the head and neck region. Some osteosarcomas contain multinucleated giant cells so plentiful that this form may be mistaken for a central giant cell granuloma.
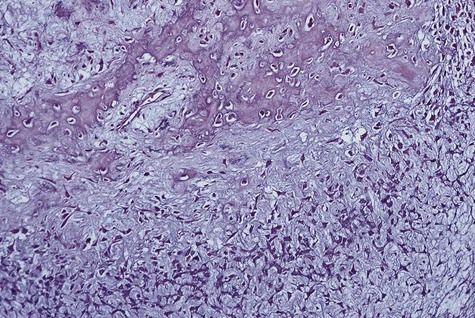
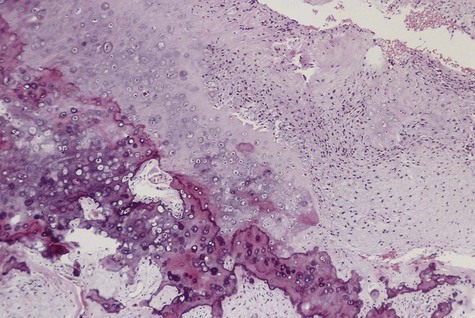
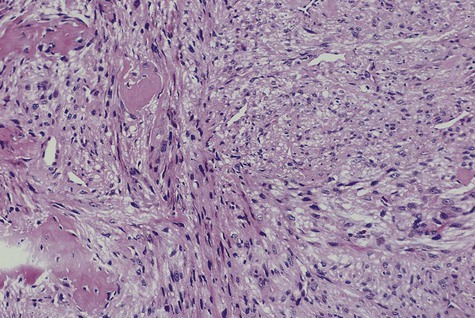
Central low-grade osteosarcoma is a rare variant that may involve the jaws. Microscopically, it resembles fibrous dysplasia because of the minimally atypical spindle cell proliferation with occasional mitotic figures and bone spicules. The microscopic diagnosis poses a challenge because of its deceptively bland features. Unlike fibrous dysplasia, the radiographic appearance is usually that of an invasive intramedullary growth with poor margination and cortical destruction. Also unlike fibrous dysplasia, the proliferation permeates bone marrow, may extend through the periosteum, and may invade soft tissues. Recurrent tumor or long-standing low-grade osteosarcoma may transform to conventional high-grade osteosarcoma (Figure 14-10).
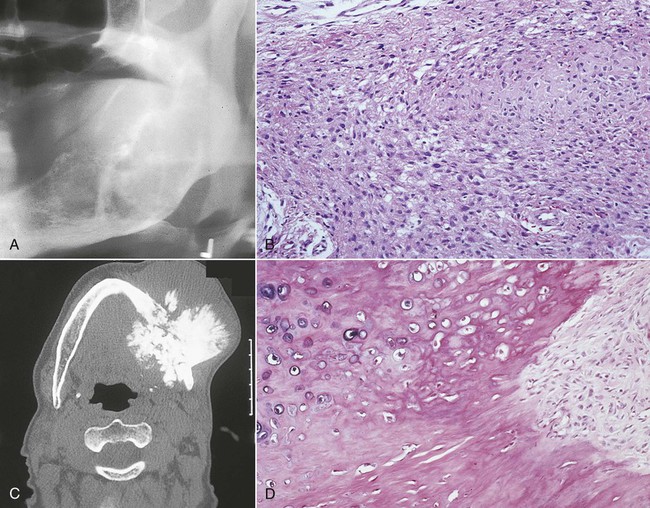
Management
Management of sarcomas of the facial skeleton involves combinations of surgery, chemotherapy, and radiotherapy. Surgical management of osteosarcoma of the mandible, however, is the mainstay of therapy and possesses numerous characteristics similar to the management of carcinoma of the jaw, with some notable differences. These similarities include required attention to surrounding anatomic barriers with their appropriate sacrifice (Figure 14-11). Invasion of anatomic barriers surrounding any head and neck tumor may be assessed by physical examination and/or special imaging studies. When a small sarcoma originates within the medullary component of the mandible, cortical bone is the first anatomic barrier the tumor encounters that forestalls its growth. Once the cortical bone is violated, the less robust periosteum is subsequently encountered. With continued growth, muscle, mucosa, and skin ultimately become invaded by the malignancy. The general approach to malignant tumor surgery of the head and neck is that at least one uninvolved anatomic barrier margin should be included on the tumor specimen as part of the en bloc resection. This practice allows better analysis of tumor margins. The main difference between resection of carcinoma in bone and resection of sarcoma lies in the recommended linear bony margin. Whereas carcinomas may be resected with a 2 cm linear margin in bone, it generally is recommended that sarcoma resections should include a 3 cm margin. Attention to proper anatomic barrier sacrifice, as well as inclusion of the recommended linear bony margin, enhances the potential for long-term palliation or cure of patients with sarcoma of the jaw.
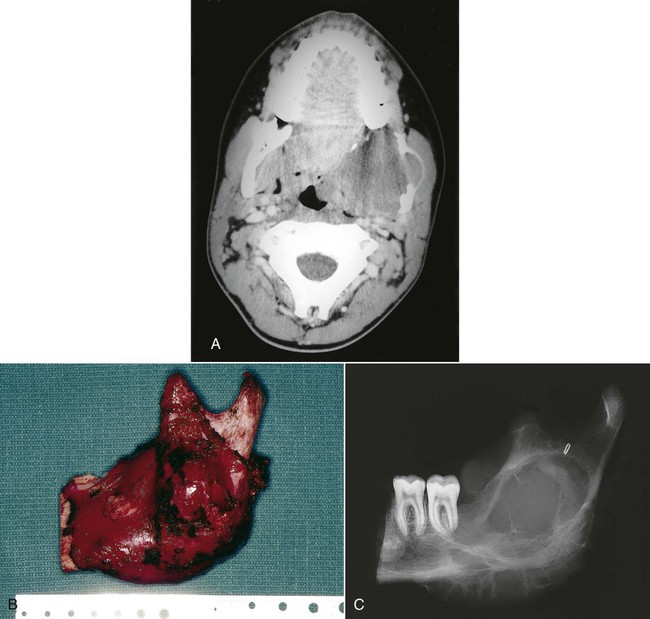
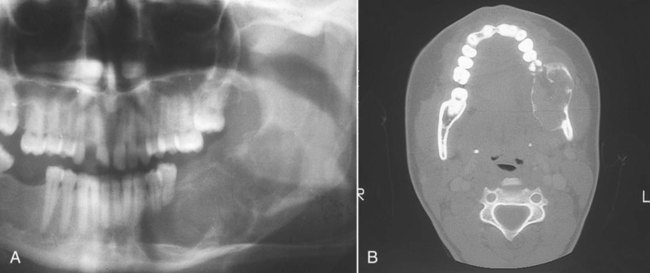
A, Axial computed tomography (CT) scan of a large fibrosarcoma of the mandible with extension into the lateral pharynx. B, Tumor resection required wide margins, including sacrifice of the condyle. Appropriate sacrifice of surrounding anatomic barriers allows for negative margins on the specimen. C, The specimen radiograph confirms the inclusion of acceptable linear bony margins with the specimen. (Reproduced with permission from Regezi JA, Sciubba JJ, Pogrel MA: Atlas of Oral and Maxillofacial Pathology. Philadelphia, 2000, WB Saunders, Figures 11-10 and 11-13.)
Parosteal Osteosarcoma
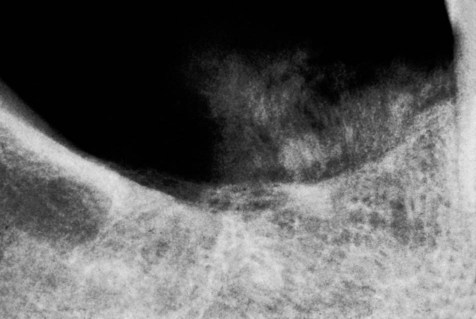
Parosteal osteosarcoma occurs over a wide age range, with a peak incidence at 39 years (Figures 14-12 and 14-13). More than 95% of cases affect the long bones, most commonly the distal femoral metaphysis, and at these sites there is a female predominance (3 to 2); when the jaws are affected, there is a male predominance. The tumor typically presents as a long-standing, slow-growing, swelling or palpable mass, often accompanied by a dull, aching sensation. Radiographically, the tumor often is radiodense (radiopaque) and is attached to the external surface of bone by a broad sessile base. It often is more radiodense at the base than at the periphery. The broad pedicle is not continuous radiographically with the underlying marrow cavity. A radiolucent clear space, corresponding to the periosteum, often can be identified between the tumor and the underlying cortex.
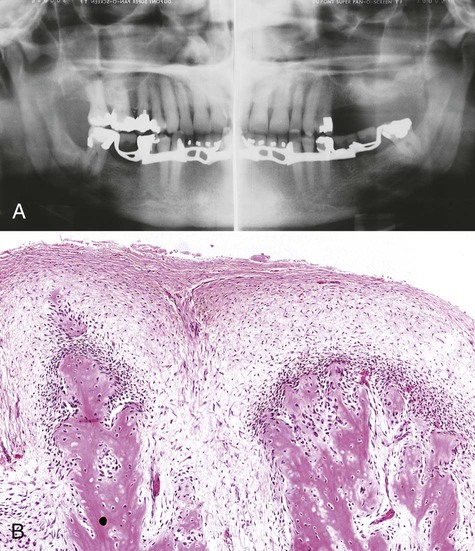
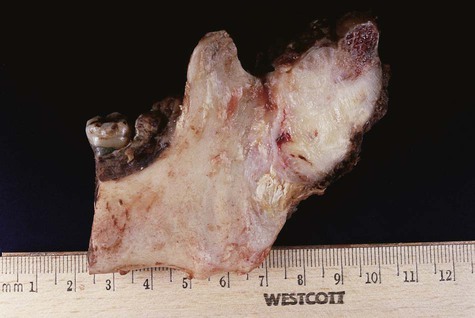
Gross specimen shows a white mass covering the ramus and condyle.
Histologically, parosteal osteosarcomas are well differentiated and are characterized by a spindle cell stroma with minimal atypia and rare mitotic figures separating irregular trabeculae of woven bone (Figure 14-14). The periphery is less ossified than the base; the lesion may have a lobulated cartilaginous cap, or it may be irregular because of linear extensions into soft tissue. Medullary involvement is unusual at initial presentation, but approximately 20% of tumors, especially recurrent ones, exhibit invasion of the underlying bone. This does not seem to affect the prognosis adversely. The bland histologic appearance of parosteal osteosarcoma raises the possibility of osteoma, osteochondroma, and exostosis.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


