Benign Nonodontogenic Tumors
Ossifying Fibroma
Ossifying fibroma is a benign neoplasm of bone that has the potential for excessive growth, bone destruction, and recurrence. It is clinically and microscopically similar, if not identical, to cementifying fibroma. Composed of a fibrous connective tissue stroma in which new bone is formed, it is classified as one of the benign fibro-osseous lesions of the jaws (Boxes 12-1 and 12-2).
Clinical Features
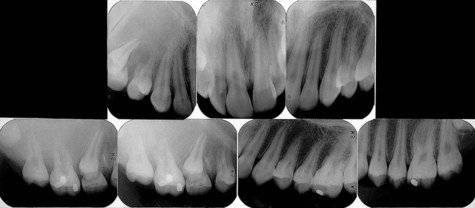
Ossifying fibroma is an uncommon lesion that tends to occur during the third and fourth decades of life, and in women more than men. It is a slow-growing, asymptomatic, and expansile lesion. In the head and neck, ossifying fibroma may be seen in the jaws and craniofacial bones. Lesions of the jaws characteristically arise in the tooth-bearing regions, most often in the mandibular premolar-molar area (Figure 12-1). The slow but persistent growth of the tumor may ultimately produce expansion and thinning of the buccal and lingual cortical plates, although perforation and mucosal ulceration are rare (Figures 12-2 and 12-3). Most of these lesions are solitary, although instances of multiple synchronous lesions have been reported; a familial background for synchronous lesions is rare.
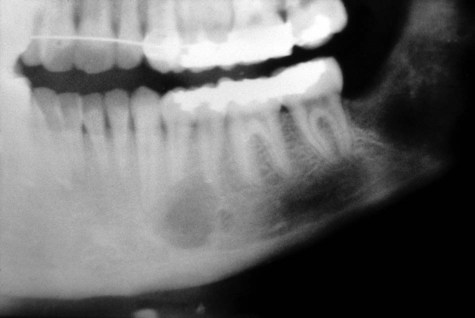
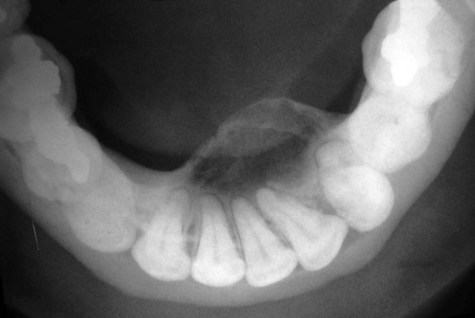
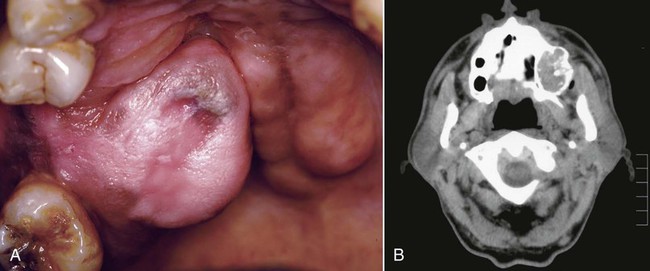
The term juvenile (aggressive) ossifying fibroma was used in the literature to describe two variants of ossifying fibroma that occur in younger patients (Box 12-4). Currently these two entities are referred to as juvenile trabecular ossifying fibroma (JTOF) and juvenile psammomatoid ossifying fibroma (JPOF). Juvenile trabecular ossifying fibroma (JTOF) typically occurs in children and adolescents; only about 20% of cases occur in those older than 15 years. The lesion occurs almost exclusively in the maxilla and mandible and rarely in extragnathic locations. JTOF is characterized by progressive and sometimes rapid growth but rarely pain. Radiographically, the tumor has a defined border and can range from radiodense to radiolucent. Microscopically, JTOF is highly cellular and contains trabeculae or spheroids of new bone. Following complete excision, recurrences of JTOF are infrequent. By contrast, the juvenile psammomatoid ossifying fibroma (JPOF) occurs principally in the extragnathic craniofacial bones, particularly the paranasal sinuses and periorbital bones, where it may cause exophthalmos, proptosis, sinusitis, and nasal symptoms. JPOF occurs in a slightly older population compared with JTOF. Microscopically, JPOF is formed by relatively cellular stroma containing small, rounded calcifications (psammomatoid). Treatment consists of surgical excision, but up to 30% of cases will show recurrences, sometimes multiple and over a span of many years.
Histopathology
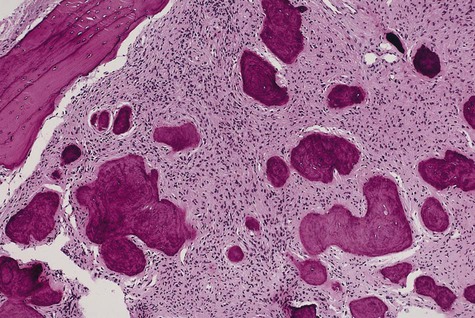
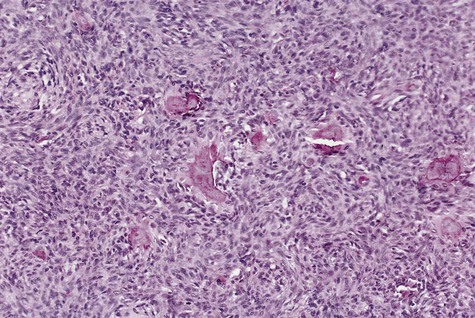
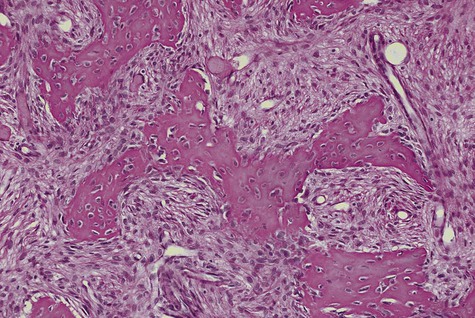
Ossifying fibroma is composed of fibrous connective tissue with well-differentiated spindled fibroblasts. Cellularity is uniform but may vary from one lesion to the next. Collagen fibers are arranged haphazardly, although a whorled, storiform pattern may be evident. Bony spheroids, trabeculae, or islands are evenly distributed throughout the fibrous stroma (Figures 12-4 to 12-6). Bone is immature and often is surrounded by osteoblasts. Osteoclasts are infrequently seen.
Fibrous Dysplasia
Fibrous dysplasia is a condition in which normal medullary bone is replaced by an abnormal fibrous connective tissue proliferation in which new, nonmaturing bone is formed (Box 12-5). A genetic defect involving Gsα protein appears to underlie this process.
Clinical Features
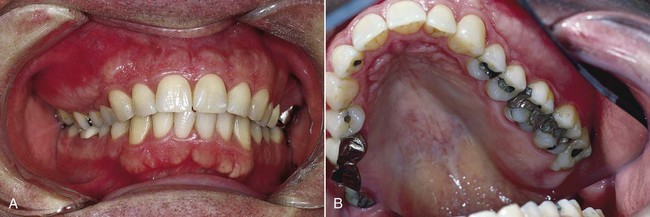
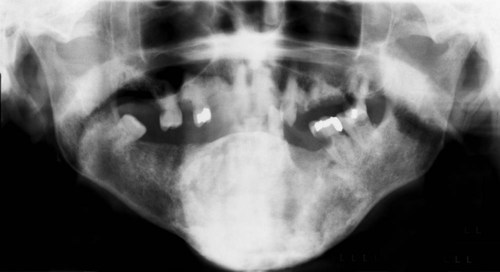
Monostotic fibrous dysplasia is much more common than the polyostotic form, accounting for as many as 80% of cases. Jaw involvement is common in this form of the disease. Other bones that are commonly affected are the ribs and the femur. Fibrous dysplasia occurs more often in the maxilla than in the mandible (Figure 12-7). Maxillary lesions may extend to involve the maxillary sinus, zygoma, sphenoid bone, and floor of the orbit. This form of the disease, with involvement of several adjacent bones, has been referred to as craniofacial fibrous dysplasia. The most common site of occurrence with mandibular involvement is in the body portion.
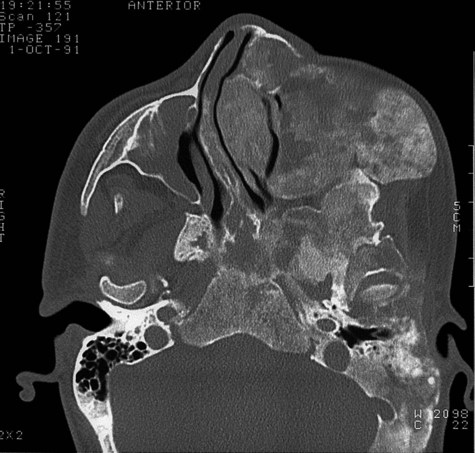
Fibrous dysplasia has a variable radiographic appearance that ranges from a radiolucent lesion to a uniformly radiopaque mass (Figures 12-8 to 12-10). The classic lesion has been described as having a radiopaque change that imparts a “ground-glass” or “peau d’orange” effect. This characteristic image, which is most identifiable on intraoral radiographs, is not, however, pathognomonic. Lesions of fibrous dysplasia may also present as unilocular or multilocular radiolucencies, especially in long bones. A third pattern, most commonly seen in patients with long-standing disease, is a mottled radiolucent and radiopaque appearance. Additional radiographic features that have been described include a fingerprint bone pattern and superior displacement of the mandibular canal in mandibular lesions.
Histopathology
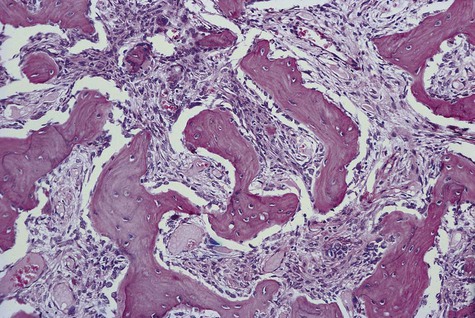
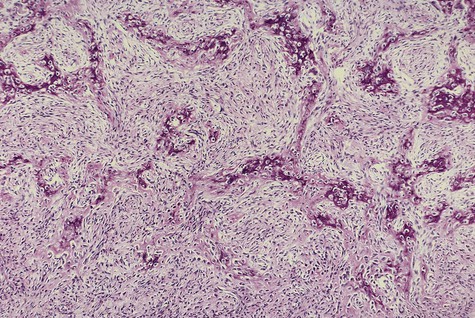
Fibrous dysplasia consists of a slightly to moderately cellular fibrous connective tissue stroma that contains foci of irregularly shaped trabeculae of immature bone (Figures 12-11 and 12-12). A relatively constant ratio of fibrous tissue to bone throughout a given lesion is characteristic. The fibroblasts exhibit uniform spindle-shaped nuclei, and mitotic figures are not seen. The bony trabeculae assume irregular shapes likened to Chinese characters (the pictograms used in Chinese writing), and they do not display any functional orientation. The bone is predominantly woven in type and appears to arise directly from the collagenous stroma without prominent osteoblastic activity. In a mature fibrous dysplasia lesion, lamellar bone may be found. Capillaries typically are prominent and uniformly distributed.
Differential Diagnosis
The primary differential consideration for fibrous dysplasia of the jaws is ossifying fibroma. As previously noted, clinical, radiographic, and microscopic features must be considered together to distinguish these processes. The well-circumscribed ossifying fibroma as compared with the diffuse fibrous dysplasia often serves as the differentiating factor. Additional features that aid in distinguishing these processes are listed in Box 12-6.
Cemento-Osseous Dysplasia
The term cemento-osseous dysplasia refers to a disease process of the jaws for which the precise cause is unknown. Cemento-osseous dysplasia describes a spectrum of disorders that include periapical cemento-osseous dysplasia, focal cemento-osseous dysplasia, and florid cemento-osseous dysplasia—apparently similar disease processes distinguished on the basis of extent of involvement of affected portions of the jaws (see Chapter 11 for a comprehensive discussion). Cemento-osseous dysplasia, ossifying fibroma, and fibrous dysplasia have been classified as fibro-osseous lesions of the jaws. These fibro-osseous diseases represent a diverse group of reactive, dysplastic, and neoplastic lesions characterized microscopically by the replacement of normal bone with a collagenous matrix containing trabeculae of immature bone and, in some instances, cementum-like material (see Boxes 12-1 and 12-2).
Osteoblastoma/Osteoid Osteoma
Osteoblastoma is an uncommon primary lesion of bone that occasionally arises in the maxilla or the mandible (Box 12-7). Osteoid osteoma is thought to represent a smaller version of the same tumor, although some prefer to separate these lesions into two distinct entities. These are benign neoplasms of undetermined cause, although a genetic defect has been suggested. Clinically and histologically, they may be confused with osteosarcoma.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


