CHAPTER 10. Genetic, metabolic and other non-neoplastic bone diseases
GENETIC DISEASES OF BONE
Osteogenesis imperfecta (brittle bone syndrome)
The term brittle bone syndrome should not be confused with the considerably more common disease of osteoporosis which is frequently termed brittle bone disease. In osteogenesis imperfecta, which is usually transmitted as an autosomal dominant trait, the bones are excessively fragile.
Genetics
The usual causal genes are COL1A1 and COL1A2. The procollagen alpha helix is defective and fails to polymerise into normal type 1 collagen and mineralise. Osteoblasts do not form bone in adequate amounts, leading to fractures. However, there is molecular and clinical diversity and types V, VI and VII disease are caused by other gene defects. Abnormal dentine (dentinogenesis imperfecta) is present, particularly in type III disease, which is associated with the COL1A2 mutation.
The bones are thin and lack the usual cortex of compact bone (Fig. 10.1), but development of epiphyseal cartilages is unimpaired so that bones can grow to their normal length. Nevertheless, they may become grossly distorted by multiple fractures and result in dwarfism. The most severe cases (type II) usually die at birth or soon after: mild cases (type V) may have little disability. In the more common form (type I), the many fractures can cause severe deformity (Figs 10.2 and 10.3). The sclera of the eyes may also appear blue because their thinness allows the pigment layer to show through (Fig. 10.4). Deafness also develops.
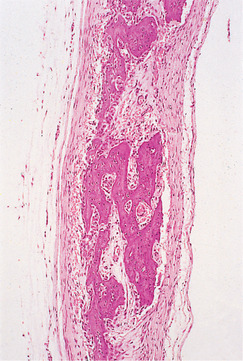 |
| Fig. 10.1
Osteogenesis imperfecta. A section from the vault of the skull of a stillborn infant with type II disease; the most severe form of osteogenesis imperfecta shows that the bone is small in amount, primitive (woven) in character and shows no attempt at differentiation into cortical plates and medullary space.
|
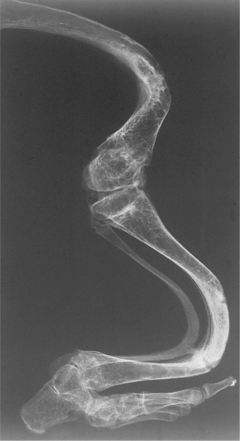 |
| Fig. 10.2
Osteogenesis imperfecta. Leg of an infant with a severe type of osteogenesis imperfecta showing severe bending as a result of multiple fractures under body weight.
|
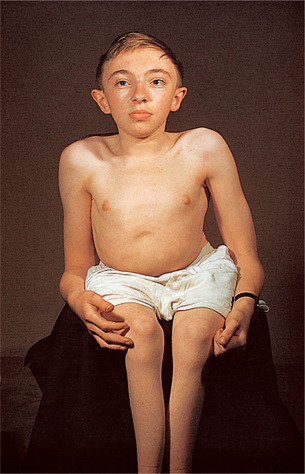 |
| Fig. 10.3
Clinical appearance of a severely affected child with osteogenesis imperfecta type III in which there is progressive deformity.
|
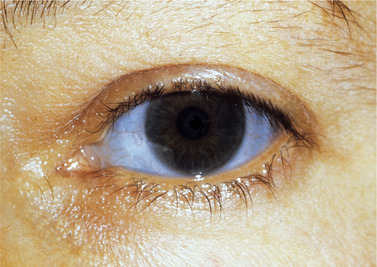 |
| Fig. 10.4
Blue sclera in osteogenesis imperfecta.
|
Adults with mild osteogenesis imperfecta account for about 25% of fractures in this disease. They also tend to suffer from back and joint pain usually due to osteoarthritis; about 60% have joint hypermobility but otherwise, the majority have good health.
Dental aspects
Obvious dentinogenesis imperfecta is sometimes associated, but all patients have some degree of abnormality of the dentine microscopically (see Ch. 2). In types III and IV, the craniofacial form is abnormal as a result of differential growth deficiency and bending of cranial structures. An abnormally ventral position of the sella region is due to bending of the cranial base and a closing mandibular growth rotation. Vertical underdevelopment of dentoalveolar structures and the condylar process are the main reasons for the relative mandibular prognathism.
Management
Success has been claimed for gene therapy using normal stromal marrow cells which can differentiate into a multiplicity of cell types including bone cells. Treatment with a bisphosphonate may be given to improve bone formation (see Ch. 6). No other treatment is effective, in which case all that can be done is to protect the child from even minor injuries and to minimise deformity by attending to fractures. Care must be taken during dental extractions, but fractures of the jaws are uncommon in this disease.
Key features are summarised in Box 10.1.
Box 10.1
Osteogenesis imperfecta: key features
• Thin fragile bones due to inadequate type I collagen formation
• Usually an autosomal dominant trait
• Autosomal recessive types (II, III, IV) – severe fragility
• Multiple fractures typically lead to gross deformities
• Dentinogenesis imperfecta associated especially in type IV
• Jaw fractures are uncommon
Osteopetrosis – marble bone disease
Osteopetrosis is a rare genetic disease in which the bones become solidified and dense (Fig. 10.5), but brittle. There is inactivity of osteoclasts and absence of normal modelling resorption. Medullary spaces are minute (Fig. 10.6) and the epiphyseal ends of the bones are club-shaped. Because of the deficiency of marrow space, the liver and spleen take on blood-cell formation, but anaemia is common and defective white cells can lead to abnormal susceptibility to infection.
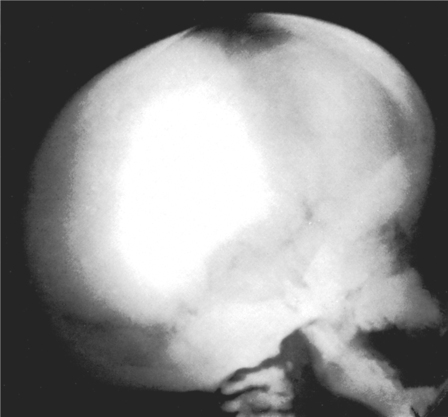 |
| Fig. 10.5
Osteopetrosis. In contrast to osteogenesis imperfecta the bone is excessively thick and dense as a result of defective resorption.
|
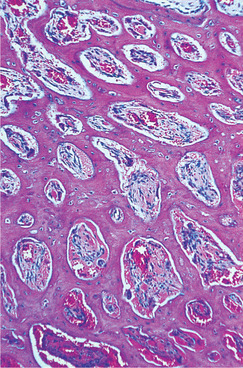 |
| Fig. 10.6
Osteopetrosis. Almost solid bone with only small lacunae has replaced the medullary space.
|
Dental aspects
There may be compression of cranial nerve canals so that trigeminal or facial nerve neuropathies can result. Despite its density, bone can be brittle so that the jaw may fracture during extractions. Osteomyelitis is a recognised complication and prevention of dental infections is important. There is no effective treatment apart from marrow transplantation.
Key features are summarised in Box 10.2.
Box 10.2
Osteopetrosis: key features
• Rare genetic defect of osteoclastic activity
• Bones lack medullary cavities but are fragile
• Extramedullary haemopoiesis in liver and spleen but anemia common
• Osteomyelitis a recognised complication
• Bone marrow transplantation offers the main hope
Achondroplasia
Achondroplasia is the most common type of genetic skeletal disorder and manifests itself as short-limbed individuals who historically became circus dwarfs. The essential defect is inability to form bone from cartilage at the epiphyses and base of the skull. This is due to a homozygous G1138A mutation at the 380 position in the fibroblast growth factor receptor 3 gene. This mutation, also termed G380R, is detectable prenatally to enable the diagnosis of the homozygous mutant type which is lethal in the newborn.
Dental aspects
While the limbs are excessively short in relation to the trunk, the head, which is normal in size, appears disproportionately large. Defective growth at the base of the skull causes the middle third of the face to be retrusive and the profile to be concave. The mandible is often protrusive and, as a result of the disparity of growth of the jaws, there is usually severe malocclusion and sometimes posterior open bite. Associated macroglossia and migratory glossitis have also been described.
There is no effective treatment, but the occlusion may be improved by orthodontic treatment.
Key features are summarised in Box 10.3.
Box 10.3
Achondroplasia: key features
• A common type of genetic dwarfism
• Failure of proliferation of cartilage in epiphyses and base of skull
• Short limbs but normal-sized skull
• Middle third of face retrusive due to deficient growth of skull base
• Malocclusion may need correction
Cleidocranial dysplasia
In this rare familial disorder, there is defective formation of the clavicles, delayed closure of fontanelles and sometimes retrusion of the maxilla. Partial or complete absence of clavicles allows the patient to bring the shoulders together in front of the chest (Fig. 10.7).
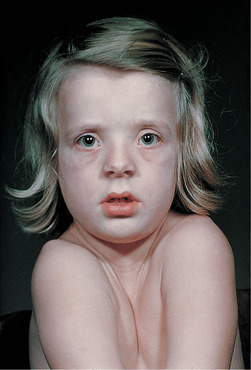 |
| Fig. 10.7
Cleidocranial dysplasia. Defective development of the clavicles allows this abnormal mobility of the shoulders. Other members of the family were also affected.
|
Dental aspects
Cleidocranial dysplasia is one of the few identifiable causes of delayed eruption of the permanent dentition. Many permanent teeth may remain embedded in the jaw (Fig. 10.8) and frequently become enveloped in dentigerous cysts (see Ch. 7). Bulging frontal, parietal and occipital bones and hypoplastic middle third of the face can produce a bird-face appearance.
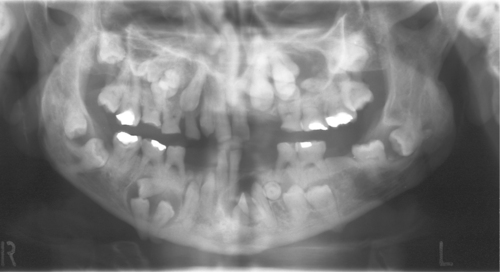 |
| Fig. 10.8
Cleidocranial dysplasia. There are many additional teeth but widespread failure of eruption and possibly development of dentigerous cysts.
|
Key features are summarised in Box 10.4.
Box 10.4
Cleidocranial dysplasia: key features
• Rare genetic disorder causing defective formation of clavicles, delayed closure of fontanelles and other defects
• Many or most permanent teeth typically remain embedded in the jaw
• Many additional unerupted teeth also present
• Sometimes many dentigerous cysts
Cherubism → Summaries pp. 136, 173
Cherubism causes multiple multilocular bone lesions in the mandible and maxilla that start in childhood, enlarge and then regress.
Cherubism is caused by one of several mutations in the SH3BP2 gene on chromosome 4p and is inherited as an autosomal dominant trait. The gene product is thought to be a signalling molecule but its exact function is unclear.
Though a dominant condition, there may be no family history because of variable expressivity and penetrance. As a result of weaker penetrance of the trait in females, the disease is approximately twice as common in males. Non-familial cases may also be new mutations. Usually, only isolated cases are encountered, but a family with no fewer than 20 affected members has been reported. The disorder may be seen almost worldwide, but appears to be rare in Japan.
The onset is typically between the ages of 6 months to 7 years, but rarely is delayed until late teenage or after puberty. Typically, symmetrical swellings are noticed at the age of 2 to 4 years in the region of the angles of the mandibles and, in severe cases, in the maxillae. The symmetrical mandibular swellings give the face an excessively chubby appearance (Fig. 10.9). The alveolar ridges are expanded and the mandibular swellings may sometimes be so gross lingually as to interfere with speech, swallowing or even breathing, but the rapidity of progress is variable. Teeth are frequently displaced and may be loosened.
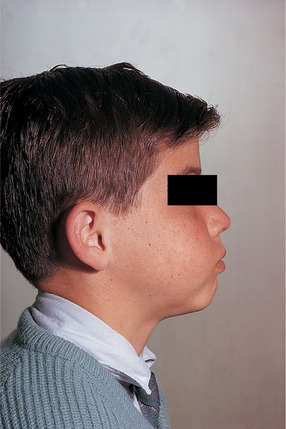 |
| Fig. 10.9
Cherubism. The typical ‘cherubic’ bulging of the cheeks can be seen.
|
Maxillary involvement is usually associated with widespread mandibular disease and is uncommon. Extensive maxillary lesions cause the eyes to appear to be turned heavenward; this together with the plumpness of the face, is the reason for these patients being likened to cherubs. The appearance of the eyes is due to such factors as the maxillary masses pushing the floors of the orbits and eyes upwards and also exposing the sclera below the pupils. Expansion of the maxillae may also cause stretching of the skin and some retraction of the lower lids. Rarely, destruction of the infra-orbital ridges weakens support for the lower lid. Maxillary involvement can also cause the palate to assume an inverted ‘V’ shape.
Despite lack of inflammation, there is frequently cervical lymphadenopathy, due to reactive hyperplasia and fibrosis. This is typically seen in the early stages and may completely subside by puberty.
Radiographic changes may be seen considerably earlier than clinical signs and are usually more extensive than the clinical swelling. The angles of the mandible are particularly involved, but the process extends towards the coronoid notch and sometimes also forwards along the body (Fig. 10.10). The lesions simulate multilocular cysts as a result of fine bony septa extending between the soft tissue masses. Panoramic radiographs help to determine the extent of the disease, but it is more clearly visualised by CT scanning.
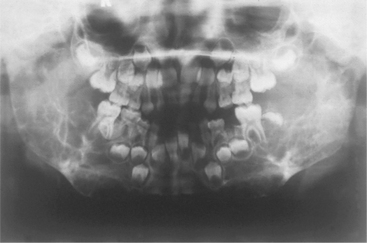 |
| Fig. 10.10
Cherubism. Both rami, much of the body of the mandible and the posterior maxillae are expanded by multilocular radiolucent lesions which have displaced and destroyed developing teeth.
|
Maxillary involvement is shown by diffuse rarefaction of the bone, but spread of lesions can cause opacity of the sinuses. A distinctive radiographic sign is exposure of the posterior part of the hard palate in lateral skull films, as a result of forward displacement of the teeth. Even after complete clinical resolution of the facial swelling, bone defects may persist radiographically. Growth is rapid for a few years, then slows down until puberty is reached. There is then slow regression until, by adulthood, normal facial contour is typically completely restored. However, radiolucent areas may persist longer. Severe cases may suffer permanent deformity, particularly if bony support for the eye has been destroyed.
Pathology
The lesions consist of multinucleate giant cells (Fig. 10.11) and resemble giant cell granulomas or hyperparathyroidism. With the passage of time, giant cells become fewer and there is bony repair of the defect (Fig. 10.12).
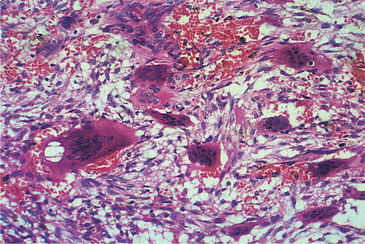 |
| Fig. 10.11
Cherubism. An early lesion showing multinucleate giant cells lying in haemorrhagic oedematous fibrous tissue. The appearances are indistinguishable histologically from giant cell granuloma.
|
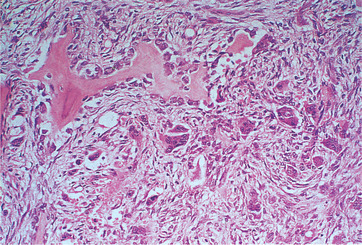 |
| Fig. 10.12
Cherubism. In a late lesion, there is formation of woven bone by the fibrous tissue and giant cells are less numerous. Eventually bone remodelling will restore the contour and quality of the bone.
|
Management
Because of natural regression of the disease, treatment can usually be avoided. If disfigurement is severe the lesions respond to curettage or to paring down of excessive tissue but, in the early stages particularly, will recur.
Key features are summarised in Box 10.5.
Box 10.5
Cherubism: key features
• Inherited as autosomal dominant trait
• Jaw swellings appear in infancy
• Angle regions of mandible affected symmetrically giving typical chubby face
• Symmetrical involvement of maxillae also in more severe cases
• Radiographically, lesions appear as multilocular cyst-like areas
• Histologically, lesions consist of giant cells in vascular connective tissue
• Lesions regress with skeletal maturation and normal facial contour restored
Hypophosphatasia
Hypophosphatasia is an uncommon recessive genetic disorder. The early-onset type causes rickets-like skeletal disease with defective mineralisation. Defective cementogenesis results in premature loss of teeth and this is sometimes the only sign of the disease (see Fig. 2.30). Plasma alkaline phosphatase levels are low, but urinary phosphoethanolamine excretion is raised. Late-onset hypophosphatasia is sometimes a dominant trait and its main manifestation is fragility of the bones.
Sickle cell anaemia and thalassaemia major
Bone changes are uncommon but, when severe, these diseases (see Ch. 25), particularly thalassaemia, can cause bony malformations of the maxillofacial region. In particular, expanded erythropoiesis causes thickening but osteoporosis of the bones of the skull. In sickle cell anaemia, infarcts in the jaws are painful. Symptomatically and radiographically, they can mimic osteomyelitis. Bone infarcts may appear relatively radiolucent at first, but become sclerotic and opaque areas in the skull or jaws are left by earlier infarcts.
In thalassaemia, the diploic spaces of the skull are enlarged due to marrow expansion and have a hair-on-end appearance radiographically and a thin cortex. The maxillae may also be expanded causing severe malocclusion. The zygomatic bones are pushed outwards and the nasal bridge depressed in severe cases.
Hyperparathyroidism–jaw tumour syndrome
This syndrome is an extremely rare autosomal dominant disease. Unlike sporadic hyperparathyroidism (see above), giant cell lesions do not develop.
Instead, cemento-osseous lesions form in the jaws. In addition to the jaw lesions, parathyroid adenomas or carcinomas, renal cysts, renal tumours and uterine tumours may be associated (see Ch. 31).
Management
The main problem is that of the parathyroid and other tumours. Parathyroid adenomas usually respond to surgery, but management of parathyroid carcinoma is more difficult. The prognosis is also affected by the behaviour of associated tumours of the kidneys or uterus.
The jaw lesions usually shell out quite readily, but some have recurred, probably as a result of incomplete excision. Otherwise, they may sometimes regress spontaneously after removal of the parathyroid tumour, though this may take months or even years.
Gigantism and acromegaly
Overproduction of pituitary growth hormone, usually by an adenoma, before the epiphyses fuse, gives rise to gigantism with overgrowth of the whole skeleton. After fusion of the epiphyses, overproduction of growth hormone gives rise to acromegaly. The main features are continued growth at the mandibular condyle, causing gross prognathism, macroglossia, thickening of the facial soft tissues and overgrowth of the hands and feet (see Ch. 31).
Fluorosis
Excessive amounts of fluorides in the drinking water, as in certain parts of Northern India, cause severe mottling of the teeth and also sclerosis of the skeleton. The intervertebral ligaments and muscle insertions calcify, causing stiffness (particularly of the back) and pain. Histologically, the bone changes are somewhat similar to those of Paget’s disease.
METABOLIC BONE DISEASE
Rickets
Vitamin D is essential for the absorption and metabolism of calcium and phosphorus. Deficiency during the period of bone development causes rickets with defective calcification and development of the skeleton, but rarely of the teeth (Ch. 30).
Defective calcium and phosphorus metabolism may also result from chronic renal disease (either hereditary or inflammatory) causing abnormal excretion of these minerals. Defective development of bone (renal rickets) can result.
The onset of rickets is usually in infancy. The main defects are broadening of the growing ends of bones and prominent costochondral junctions due to the epiphyseal defects (Fig. 10.13). The weakened bones bend readily. Typical changes in the skulls are wide fontanelles, bossing of the frontal and parietal eminences and thinning of the back of the skull. Radiographs show the wide, thick epiphyses and deformities (Fig. 10.13). There are usually normal serum calcium but low phosphorus levels, or either may be depressed. The alkaline phosphatase level is raised.
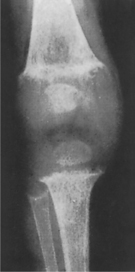 |
| Fig. 10.13
Rickets. Overgrowth of cartilage (as shown in the section in Figure 10.14) causes the epiphyseal plate to be broad, thick and irregular, and the ends of the bone become splayed. The growing end of the bone is ill-defined and calcification defective.
|
Pathology
Throughout the bone, but especially at the ends of the shafts, trabeculae become surrounded by newly formed, uncalcified osteoid matrix. In the zone of provisional calcification, mineralisation of cartilage fails and the cartilage cells continue to proliferate until the epiphyseal plate becomes greatly thickened, wide and highly cellular (Fig. 10.14). Blood vessels invade and branch irregularly among the proliferating cartilage cells. They are accompanied by connective tissue which further disorganises the epiphysis.
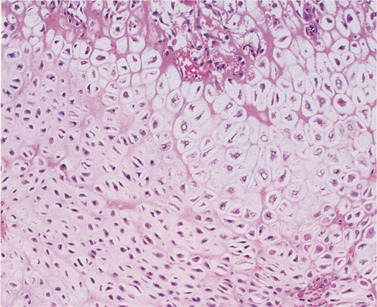 |
| Fig. 10.14
Rickets. Microscopically, the epiphyseal plate has become disorganised with loss of the well-drilled lines of chondrocytes. There is also fibrous proliferation, but no calcification.
|
Dental aspects
Teeth have priority over the skeleton for minerals and dental defects rarely result from rickets. Hypocalcification of dentine, with a wide band of predentine and excessive interglobular spaces, may be seen in unusually severe rickets. Eruption of teeth may also be delayed in such cases. Rachitic children are not abnormally susceptible to dental caries and ‘shortage of calcium’ as a cause of dental caries is a myth.
Treatment
Vitamin D, the equivalent of 2000 to 3000 i.u. daily, should be given. The diet should also be adequate in other components. Orthopaedic treatment may be needed to correct deformities.
Vitamin D-resistant rickets
This term is given to familial hypophosphataemia. The skull sutures may be wide but the dental changes are more striking. The pulp chambers are abnormally large and calcification of dentine is defective. Pulpitis, often due to minimal caries, and apparently spontaneous dental abscesses are typical complications. Preventive dental care using fissure sealants and other means is therefore essential.
Scurvy
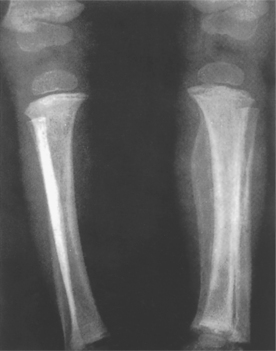
Scurvy, caused by vitamin C deficiency, leads to defective formation of collagen and osteoid matrix; such matrix as forms is well calcified. Infantile scurvy, with skeletal defects, is largely of historical interest in Britain but has been described, for instance, in an infant whose mother had eccentric ideas about diet. A case with gingival bleeding was described in 2000.
The amount of osteoid matrix formed is small but highly calcified so that the ends of the long bones are sharply defined radiologically. Weakness of the connective tissue and the haemorrhagic tendency (purpura) cause detachment of the periosteum by bleeding and bone pain. The haematoma becomes calcified as shown in the radiograph (Fig. 10.15).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


