This review addresses the multicausal etiology of periodontitis, in which genetic factors play a role. The various proposed causes for periodontitis always work simultaneously, but the relative contribution of each of these varies from case to case. We are still at an early stage to identify the genes involved, in comparison with other chronic diseases. To date, the genetic variations firmly and repeatedly associated with periodontitis in some populations are found within the following genes: ANRIL, COX2, IL1, IL10, DEFB1, whereas many other proposed periodontitis candidate genes have not been firmly proven or replicated.
Key points
- •
Periodontitis is a multicausal disease, with each of the causal factors playing a role but the relative contribution of these vary from case to case.
- •
The disease behaves in a nonlinear fashion, with periods of aberrant host response and periods of a disease resolving state.
- •
To date only a few of the multitude of possible genetic factors for periodontitis have been identified.
Introduction
Periodontitis is a chronic inflammatory disease of the supporting tissues around the teeth, which results in irreversible periodontal attachment loss, alveolar bone destruction, subsequent tooth mobility, and, ultimately, if left untreated, tooth exfoliation. There are 2 main types of periodontitis: aggressive periodontitis (AgP) and chronic periodontitis (CP). Severe periodontitis occurs in approximately 8% to 15% of the population depending on the definitions used for severe periodontitis and depending on the specific study population subjected to epidemiologic studies. In countries with a high availability of dental care, with dental and health awareness, and with preventive measures available, the prevalence of severe periodontitis may be less than 10%. In contrast, the prevalence can even be greater than 15% in less developed countries with no dental care. Recent studies suggest that almost half of the population suffers from mild to moderate periodontitis. Nevertheless, severe periodontitis is a disease occurring only in a minority of the population (8%–15%) and specific susceptibility factors play a role.
This article discusses the multicausal etiology and complexity of periodontitis with emphasis on the genetic risk factors. It is based in part on a recent review.
Introduction
Periodontitis is a chronic inflammatory disease of the supporting tissues around the teeth, which results in irreversible periodontal attachment loss, alveolar bone destruction, subsequent tooth mobility, and, ultimately, if left untreated, tooth exfoliation. There are 2 main types of periodontitis: aggressive periodontitis (AgP) and chronic periodontitis (CP). Severe periodontitis occurs in approximately 8% to 15% of the population depending on the definitions used for severe periodontitis and depending on the specific study population subjected to epidemiologic studies. In countries with a high availability of dental care, with dental and health awareness, and with preventive measures available, the prevalence of severe periodontitis may be less than 10%. In contrast, the prevalence can even be greater than 15% in less developed countries with no dental care. Recent studies suggest that almost half of the population suffers from mild to moderate periodontitis. Nevertheless, severe periodontitis is a disease occurring only in a minority of the population (8%–15%) and specific susceptibility factors play a role.
This article discusses the multicausal etiology and complexity of periodontitis with emphasis on the genetic risk factors. It is based in part on a recent review.
Periodontitis is a complex disease
The current concept of the etiology of periodontitis is that it is a complex disease . Complexity of periodontitis means that it is a process involving multiple causal components, which interplay with each other simultaneously. Complex systems are almost always nonlinear . Nonlinearity in complex systems means that the causes and effects are disproportional so that a small cause may result in a large effect or a large cause may result in a small effect, and that the disease progression rate fluctuates, or rather, can move from one state to another and back. Nonlinearity in periodontitis is revealed by heterogeneity in its clinical course; the latter is a common clinical finding by the many periodontal specialists who treat patients with periodontitis.
There are several main causal risk factors: (1) the subgingival bacterial biofilm on both the tooth root surface and on the pocket epithelial lining ; (2) genetic risk factors and epigenetic modifications ; (3) lifestyle–related risk factors, such as smoking, stress and poor diet ; (4) systemic disease, notably diabetes ; and (5) other as of yet unknown factors (eg, occlusal disturbances or fremitus, iatrogenic causes) ( Fig. 1 ).
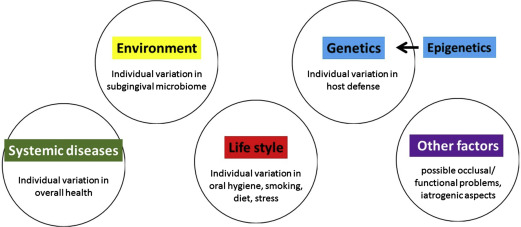
The 5 main causal components for periodontitis can be brought together into a pie chart. Fig. 2 presents a generic multicausality model for periodontitis, where each of the 5 causal components have an equal contribution.
However, it is important to note that the relative contribution of each of the causal factors varies from patient to patient ( Fig. 3 ). In general, older patients with CP are considered to have a major contribution from environmental and lifestyle factors. So, many years of biofilm accumulation and unfavorable lifestyle behaviors like smoking, poor diet, and no or irregular visits to dental professionals, likely make substantial contributions to disease progression (see Fig. 3 ). On the other hand, periodontitis in younger patients, for example suffering from AgP, can be caused to a greater extent by genetic factors (see Fig. 3 ).
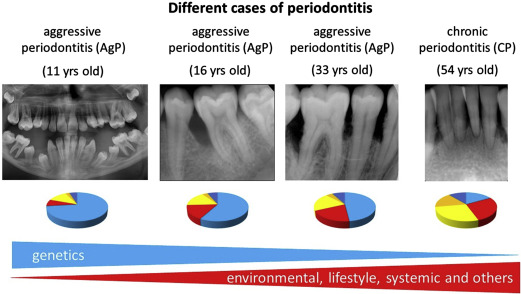
Evidence for the role of genetics in periodontitis has been gained from population, family, and twin studies. Studies in twins and especially in monozygotic twins are a strong and preferred model to study heritability of a disease. Michalowicz and colleagues studied 110 twin pairs (63 monozygotic and 33 dizygotic twin pairs) with CP and showed that among monozygotic twins a higher concordance of alveolar bone loss patterns was seen than in the dizygotic twins. The heritability estimates for alveolar bone loss among reared-apart monozygotic twins was 38%. For the early-onset form of periodontitis, often equated with AgP, there is only 1 study that has reported on the heritability in twins. The study consisted of a low number of twins (7 monozygotic and 19 dizygotic twin pairs at age 12–17 years) and therefore no clear conclusions could be made on the concordance rate of early-onset periodontitis. Because of a relatively low prevalence of AgP in the general population, it is very difficult to identify enough affected twins to provide sufficient statistical power to test the concordance of this disease phenotype. Nevertheless, because of strong familial aggregation, rapid progression, and early onset of disease, it is clear that genetic factors play a large role in the disease susceptibility of AgP (see Fig. 3 ). Thus, the earlier periodontitis manifests itself, the greater the role of genetic factors, which is similar to other complex inflammatory diseases.
Periodontitis development and progression
The bacteria in the subgingival biofilms, as well as their toxic and antigenic products (eg, endotoxin, bacterial metabolic components), initiate the inflammatory reactions that recruit polymorphonuclear neutrophilic granulocytes (PMN) and other inflammatory cells into the gingival tissues. Subsequently, the recruited immune cells, in particular PMNs, but also activated pocket epithelial cells and fibroblasts in the underlying matrix, release proinflammatory mediators, including cytokines, prostanoids, and proteolytic enzymes.
The type and severity of the periodontal inflammatory reaction to the dental biofilm is determined by genetic risk factors and the other causal factors named previously. The disease-type of inflammatory reactions in the periodontal tissues, results in a white-cell infiltrated connective tissue, loss of periodontal ligament, and resorption of alveolar bone. This destruction is thought to be “collateral damage” due to the inflammatory processes. Collagen and intercellular matrix degradation is thought to be caused by the activity of a large variety of proteolytic enzymes derived from fibroblasts and PMN. Moreover, enhanced osteoclastogenesis and osteoclast activation is initiated by cytokines and prostanoids, like interleukin (IL)-1 and prostaglandin (PG)-E2 respectively, resulting in alveolar bone destruction.
The host response (ie, the inflammatory reactions) varies from person to person, and is determined by genetics and other factors known to lower host responses, such as an unfavorable lifestyle and/or systemic disease such as diabetes (gene–lifestyle interactions). The genetic blueprint of an individual is not a black and white deterministic causal factor. It is very dependent on the type and function of variants in multiple genetic loci: (1) multiple genes play a role (polygenic) (gene-gene interactions) and (2) not all cases have the same genetic risk variants and (3) multiple, genetic variations are present at the same time. All these genetic variations essentially modify the host response, which is also not set at a constant level. The host response can fluctuate or swing between states. The patient’s susceptibility to periodontitis (and many other complex chronic diseases), is determined by the complex interplay between the environment (ie, bacteria) and the host. Here, genetic variations may cause modifications in the immune system, and the immune system is also affected by lifestyle factors such as smoking, stress, and diet, as well as certain systemic diseases (eg, diabetes) (see Fig. 1 ). Furthermore, epigenetic changes of DNA and mutations during lifetime may modify an individual’s susceptibility to periodontitis.
A depiction of the complex system of periodontitis can be attempted on the basis of the current understanding and interpretation of complexity. Undisturbed subgingival biofilms become maximum climax pathogenic communities as described in the literature, which are considered a state of stable equilibrium. In mathematical terms, the bacteria of the subgingival biofilms come into contact with the host defense elements through random interactions and beyond a criticality in their interaction, the confrontation of bacteria with the host defense can be driven out of equilibrium. Interestingly, it has been proposed that there are keystone pathogens that elevate community virulence and the resulting dysbiotic community targets specific aspects of host immunity promoting an aberrant immune response. At this point, self-organization (SO) of the host defense system occurs. SO represents a spontaneous arrangement of the system from a state of initially separated parts to a final state of joined and tied-up parts. As the term implies, SO is a complex system’s evolutionary process without any external factor imposed. SO happens in a space of scale invariance. Such a space is provided by periodontal ligament, which is a scale invariant self-similar object. SO is based on (1) amplification of disease activity by positive feedback, (2) balancing of disease activity by negative feedback, (3) amplification of random fluctuations (all 3 situations are obvious causes of nonlinearity), and (4) on multiple local interactions of the system’s components. In other words, periodontitis can fluctuate or move between an aberrant immunologic reaction state and a resolving state (resolution).
Fluctuations and chance are parts of a complex system’s behavior (mathematical term for host response) and random perturbations (“noise”) facilitate the system to make a series of attempts to explore all possibilities and finally make a choice and stabilize at a state for some period; this can be regarded as an order out of “noise.” A coherent global behavior appears as SO (eg, the system behaves as AgP [aberrant immune reactions] or CP [minor disease progression or in resolving state, often no differences in immune reactions found between CP cases and healthy controls]). The resultant emergent global behavior offers the ground for predictability out of the system’s chaotic (aperiodic, sensitive to initial conditions and therefore unpredictable) behavior. After SO occurrence we can no longer reduce the description of the system to one of its parts: all contributing parts (eg, genetics, environment, lifestyle) are tied up into a collective host defense system.
It is suggested in the literature that even the immune response level mounted at the early stage of gingivitis is the determinant factor of periodontitis progression and not the presence of specific bacteria known for their virulent properties. The host response level can vary widely without affecting the SO, and there is no real specific value for the host response level for SO to occur.
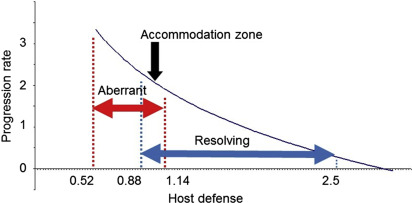
Thus, we can formulate the hypothesis that the host response against the subgingival microbiome is a function of host genetics, environmental and lifestyle factors, systemic disease(s), and unknown factors. However, the outcome is not simply a summation. There are no clear-cut borders in the behavior of complex systems; there is always a mixture of regularity, chance, and fluctuations. In most of the population, the host response is basically “normal” regarding the susceptibility to periodontitis. The host response swings like a pendulum, from the aberrant immune response to the resolving state, and it always passes through a settlement or accommodation zone with little disease progression ( Fig. 4 ), and where the level of the host response is essentially normal, not different from immune parameters in healthy controls. In the light of recent epidemiologic data that point that periodontitis is a pandemic disease, we propose the paradigm that the host response is mostly normal and comparable with healthy controls. However, as the system is always in some sort movement, it can swing into an aberrant immune function (one can observe this as an AgP case) or it can swing into a resolving or resolution phase (slow periodontal progression of many CP cases). Both the aberrant and resolution-type host responses for periodontitis have been suggested in the literature.
Some theoretic background on genetics
Hundreds of thousands genetic loci can be identified in the human genome (ie, the complete set of chromosomes). These are stretches of DNA on the chromosomes with an order, with a start and an end. Many of these genetic regions do not contain a gene proper (a genetic region coding for a protein), rather just regulatory elements or DNA with unknown function (originally called “junk DNA”). There are some 20,000 to 30,000 genetic loci that do contain one or several genes coding for proteins and having flanking regulatory regions.
Genes direct the production of proteins with the assistance of enzymes and messenger molecules. In humans, the genes are located on 23 pairs of chromosomes, 22 pairs of autosomal chromosomes and 1 pair of sex chromosomes (XX for females and XY for males). From each pair, one chromosome is inherited from the father and one from the mother.
In the chromosomes, DNA is arranged in a double helix: 2 polynucleotide chains are associated together by hydrogen bonding between the nitrogenous bases. The pairing of the 2 single-stranded nucleotide chains is complementary: G pairs only with C, and A pairs only with T; these are called base pairs (bp). The sequences of these 4 nucleotides determines the information encoded in the DNA. Every cell in the body contains a complete copy of the approximately 3 billion DNA base pairs that make up the genome.
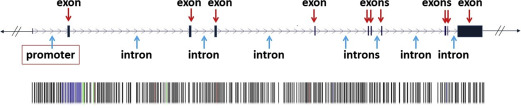
A genetic locus containing a gene is illustrated in Fig. 5 . This genetic locus consists of various parts. A promoter region is a sequence of nucleotides upstream of the coding region that contains specific sequences of nucleotides that are essential for the regulation and initiation or inhibition of the transcription of the coding region. Introns are often relatively large stretches of non–protein coding nucleotides within the gene, which also often participate in the regulation of transcription of DNA into mRNA. Another function of introns may be to help in splicing events of mRNA before the translation step. Introns are surrounded by exons , which code for the sequence of amino acids of a protein. For transcription and translation into a protein, the genetic code in such an exon is read in groups of 3 nucleotides; each trinucleotide sequence (triplet) is called a codon, which encodes a specific amino acid. The collection of known exons in our genome is called the exome .
Genes can be transcribed in alternative ways, where the regulatory regions play decisive roles; thus with this, each of the estimated 20,000 to 30,000 genes in the human genome code for an average of 3 proteins. Proteins make up all the body structures, and of course are essential for development and function of teeth, oral mucosa, and the periodontium. Proteins also carry signals between cells, code for components of the immune system, serve as enzymes, and control biochemical reactions. If a cell’s DNA is mutated, an abnormal protein or abnormal quantities of protein may be produced, which can disrupt the body’s usual processes and lead to a disease.
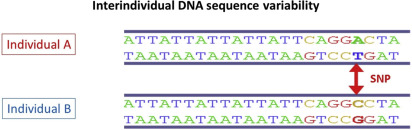
Nucleotide sequencing technologies determined the exact orders of the base pairs in the DNA. The international Human Genome Project finished the first working draft sequence of the entire human genome in 2000, and a first high-quality reference sequence of the human genome was successfully completed in 2003. In May 2006, the finished high-quality version of the sequences of all human chromosomes was published. It showed that the genomes of any 2 people are more than 99% identical, but variations between the individual genomes exist and differ on average in about 1 in every 1200 bp. Differences in individual bases are by far the most common type of genetic variation, and are known as single nucleotide polymorphisms (SNPs ) (see Fig. 5 ; Fig. 6 ).
How to identify periodontitis-associated genetic markers
Candidate Gene Approach
In general, the genetic loci and the known genetic polymorphisms occurring in such loci, are chosen for candidate gene association studies in periodontitis based on their relation with immune responses and/or have previously been associated with other chronic inflammatory diseases, such as rheumatoid arthritis, cardiovascular disease, Crohn disease (CD), type 2 diabetes, and systemic lupus erythematosus. The overlapping genetic risk factors for common chronic inflammatory diseases is called pleiotropy .
Approximately 11 million SNPs are estimated to occur commonly in the human genome. All known common SNPs are listed in the catalog of common genetic variation, the HapMap, which was generated by the HapMap project and first published in 2005. It describes the characteristics of the variants, where they occur in the DNA, and how they are distributed among people within populations and among populations ( www.hapmap.org ).
The catalog with the highest coverage of human genetic variation, obtained from population-based sequencing was published in 2010 by the “1000 Genomes Project” ( www.1000genomes.org ), and reported that each individual differs by more than 11 million often very rare SNPs, and that in each generation greater than 30 de novo mutations arise per individual.
The different variants at a specific genetic position are called alleles , and the individual’s personal unique collection of alleles in his or her genome makes up the individual’s genotype . Two or more alleles for a given position may exist in nature, and occur with different frequencies. The minor allele frequency (MAF) in a population is the proportion of the least frequent allele at a given chromosomal/genetic position and can range from 0% to 50%. Variants with an MAF greater than 5% in a population are termed common variants. If the MAF of a variant ranges between 1% and 5% it is called a rare variant. Genetic variants with frequencies less than 1% are called mutations.
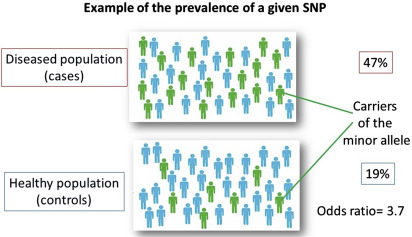
In candidate gene association studies, allele frequencies of selected SNPs are determined and compared between a well-defined group of cases and controls ( Fig. 7 ). Next to the statistical significance, the results are expressed with an odds ratio (OR). The ORs of common gene polymorphisms associated with complex diseases are typically less than or equal to 1.5 (in Fig. 7 for the purpose of illustration an extreme OR of 3.7 is shown).
A mutation or a genetic variant may have no effects on phenotype, or may have moderate to strong effects. For example, if a transition has taken place within the coding region of a gene, it may result in an amino acid substitution and therefore an altered protein structure, which may affect its function (nonsynonymous SNP). Or, when such mutations have taken place in the promoter region of the gene, it may alter the expression levels of the gene. Accordingly, genotypic differences among individuals can contribute to phenotypic variation, which is termed the genetic variance. How strongly a genetic variant affects the susceptibility to a disease is defined as the genotype relative risk, the ratio of the risk of disease between individuals with and without the genotype. A ratio of 1.1 equates to a 10% increase in risk and is often expressed as the OR. However, carriership of a genetic variant or mutation does not inevitably lead to disease, but only a proportion of individuals with a mutation or risk variant will develop the disease, this is known as penetrance . The severity of the disease in individuals who carry the risk variant and actually have disease, is described by the expressivity of the variant. As explained previously, factors other than genetic risk factors also must play a role in disease expression.
Despite the existence of many genetic variants, only a fraction of the genotypic differences contributes to phenotypic variation. The exact locations where in the chromosomes the true causative variants are located is mostly unknown. Testing all of the several millions of common and rare SNPs in a person’s chromosomes would be extremely expensive. But variants that are near each other tend to be inherited together (eg, people who have a nucleotide A rather than a nucleotide G at a particular location in the chromosome can have identical genetic variants at other locations in the chromosomal region surrounding the A ). This nonrandom association between alleles at different loci is termed linkage disequilibrium (LD) and the regions of linked variants are known as haplotypes ( www.hapmap.ncbi.nlm.nih.gov ). Determining the identity of a common SNP in a haplotype, the tag SNP, uniquely identifies all other linked variants on the same haplotype. Testing an individual’s tag SNPs, enables the identification of haplotypes in the chromosomes. If patients tend to share a particular haplotype, variants contributing to the disease might be somewhere within or near that haplotype. The number of tag SNPs that contain most of the information about the patterns of genetic variation of a genome is estimated to be 300,000 to 600,000, which is far fewer than 11 million common SNPs, and extremely less expensive to genotype. Thus, the information from the HapMap has been instrumental to map variants associated with various diseases.
The genetic factors associated with or actually contributing to the pathogenesis of periodontitis have been identified to a limited extent. In the past decades research on the genetics of periodontitis has focused on identifying specific SNPs in specific genetic loci as risk factors for AgP and CP. In general, genetic studies using a severe disease phenotype are the most useful to identify the genes involved in the disease. For complex diseases, the strongest phenotypes will be affected most by genetic factors (genetic penetrance is high) and will suffer the least from “noise” from environmental, lifestyle, systemic, and other factors (see Figs. 1–3 ); in other words, there is more genetic homogeneity in such case populations. Therefore studies using AgP study populations may have less genetic heterogeneity than CP study populations. The genetic risk loci identified in AgP can then be carried forward for testing as candidate risk loci in CP. Notably, as stressed previously, the SNPs identified for periodontitis mostly point to a gene locus of importance, where these SNPs serve only as genetic markers and not particularly as the causative variants.
A large number of candidate gene association studies have been performed in periodontitis with varying and often contradictory results. Small cohort size resulting in lack of power and the lack of replication have been the major problems for most periodontal genetic studies. Further, most of the candidate gene studies in periodontitis have not captured the complete genetic information of a particular region of interest. In almost all studies, only one or a few candidate SNPs instead of complete haplotypes of the genetic locus of interest were genotyped. Furthermore, many studies on genetics in periodontitis are limited because of inadequate phenotype classification of periodontitis and control subjects, as well as not taking into account lifestyle factors, such as smoking, possible differences in allele carriership related to gender, or the presence of other diseases.
The Genomewide Association Study Approach
A genomewide association study (GWAS) is a powerful molecular technique to analyze hundreds of thousands or even millions of variations in genomic DNA simultaneously and to determine if any genetic locus is associated with a certain disease phenotype. It is an open-ended or hypothesis-free approach; no a priori genetic candidate is being investigated. GWAS analyze SNPs covering the entire human genome with the great majority in regulatory regions and a small part in coding regions. Because of the complex structure of the genome (for example LD), associated variants are mostly not the causative variants; however, the associated variants that are identified are expected to be linked with the causative variants. This can be uncovered in subsequent genetic fine mapping experiments.
Because in a GWAS an enormous number of SNPs are determined, it is essential to include large numbers of well-characterized (most preferably strong phenotypes, such as AgP rather than CP) homogeneous patient populations (all the same ethnic background) and as good as possible, matched controls. Importantly, the statistical significance level for P is usually set at less than 5 × 10 −8 to correct for extensive multiple testing. Typically, on the basis of a GWAS, novel candidate genetic loci or SNPs can be proposed; importantly, the results of a GWAS need to be further validated by an independent cohort with a candidate gene study. Therefore, GWAS are instrumental in discovering novel candidate genes and their possible roles in biological pathways, especially in diseases in which the genetic basis is not understood.
Today, more than 1300 GWAS have been performed ( www.ebi.ac.uk/gwas/ ), but only 5 GWAS have been performed in association with periodontitis; 1 in AgP and 4 in CP. Another GWAS in periodontitis studied SNPs in association with pathologic periodontal pocket depths, rather than in association with a certain form of periodontitis. Furthermore, 1 GWAS examined host genetic risk loci in association with subgingival bacterial colonization. Recently, Rhodin and colleagues reanalyzed the GWAS data of previous studies, with a gene-centric and gene set enrichment analyses. We report the validated gene variants associated with periodontitis from these GWAS studies in combination with candidate gene studies later in this article. Notably, the studies differ in methodology and quality, and level of validation.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


